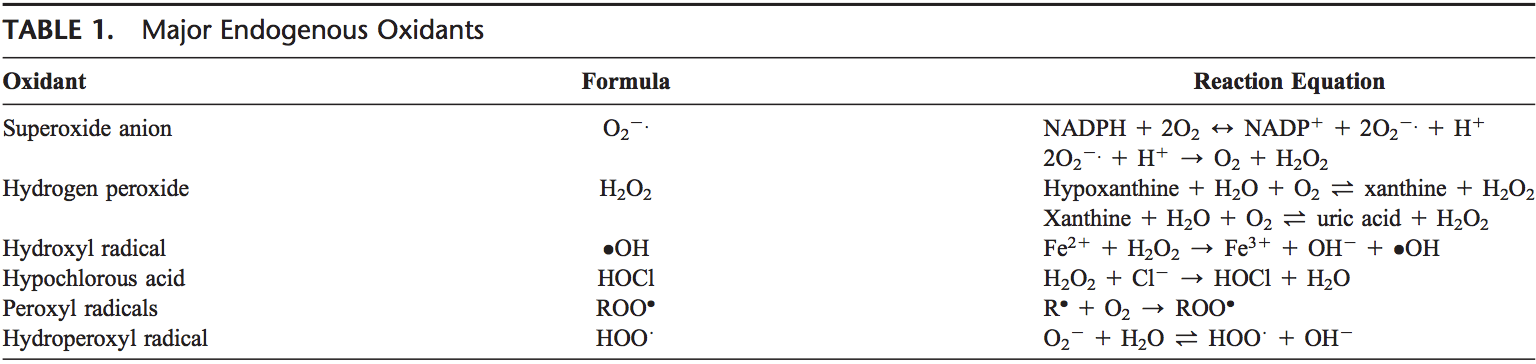
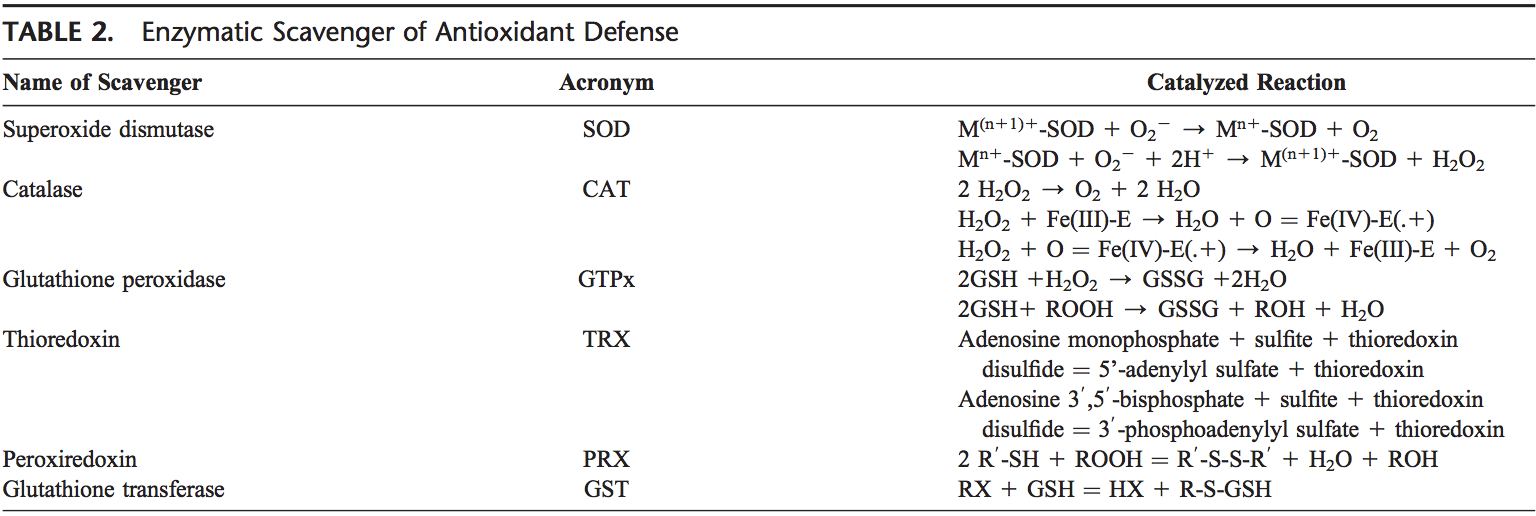
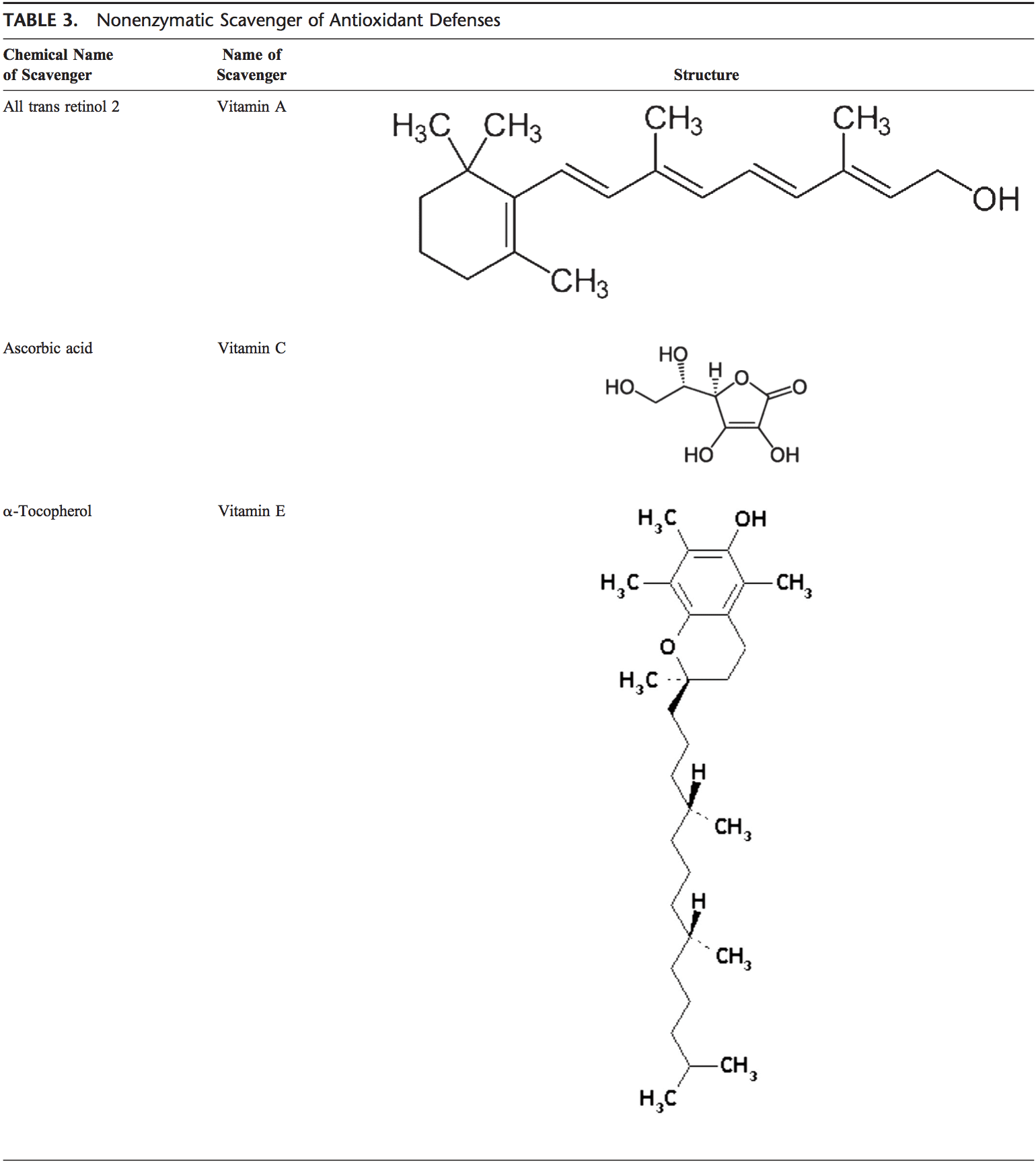
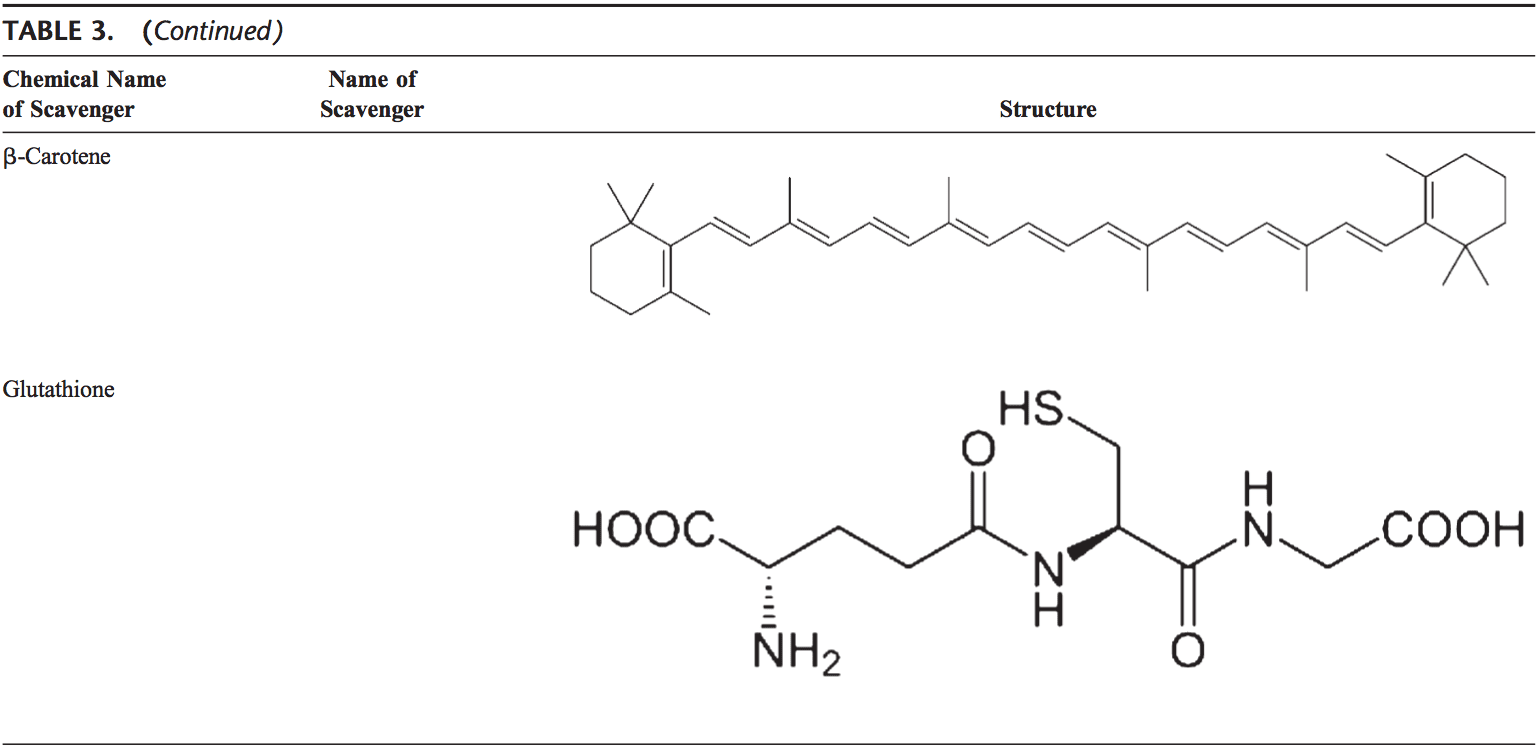
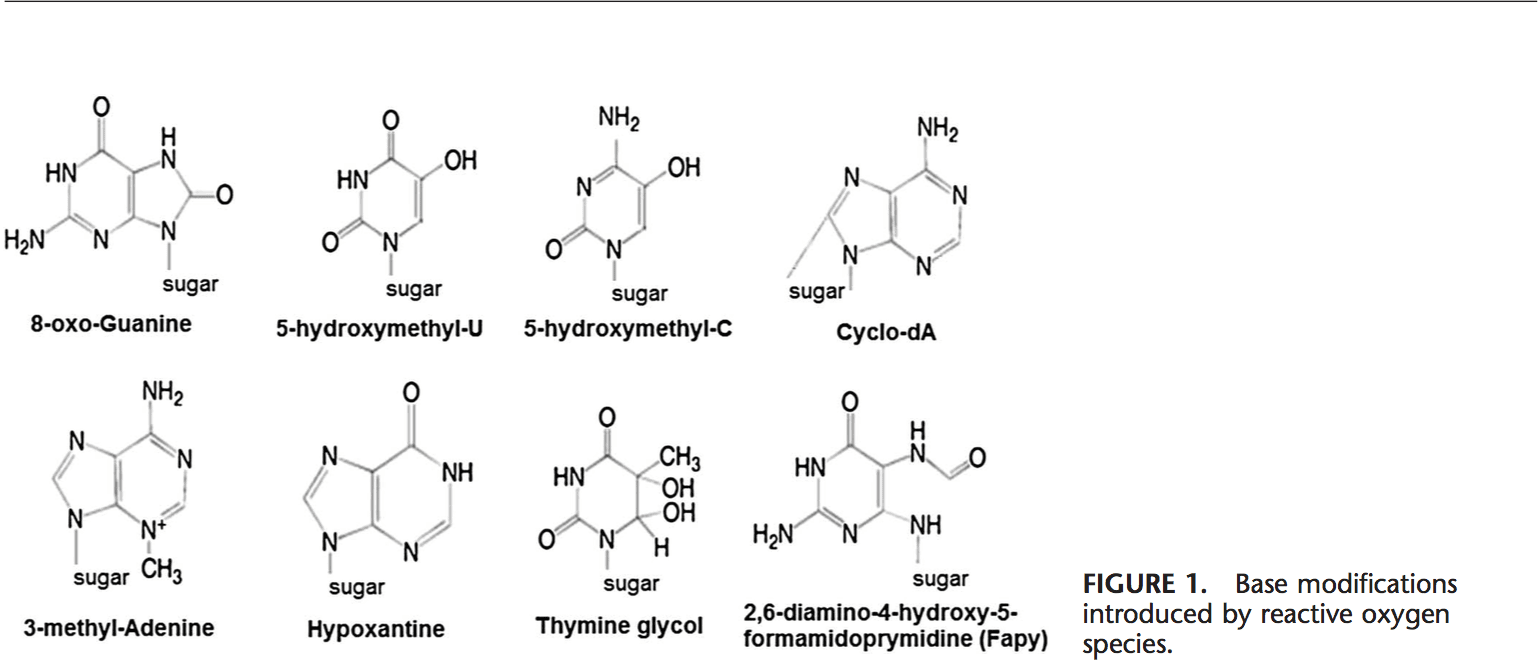
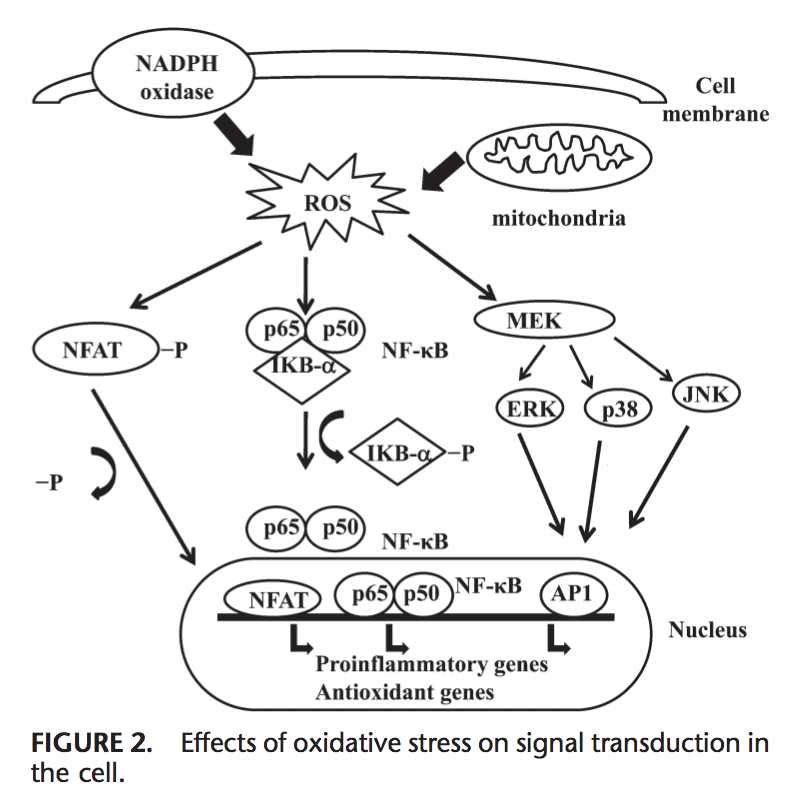
Voltar Clinic Oxidative Stress Quiropraxia e Equipe de Medicina Funcional. O estresse oxidativo é definido como um distúrbio no equilíbrio entre a produção de oxigênio reativo (radicais livres) e as defesas antioxidantes. Em outras palavras, é um desequilíbrio entre a produção de radicais livres e a capacidade do corpo de neutralizar ou desintoxicar os efeitos nocivos por meio da neutralização por antioxidantes. O estresse oxidativo leva a muitas condições fisiopatológicas no corpo. Estes incluem doenças neurodegenerativas, ou seja, doença de Parkinson, doença de Alzheimer, mutações genéticas, cânceres, síndrome da fadiga crônica, síndrome do X frágil, distúrbios cardíacos e dos vasos sanguíneos, aterosclerose, insuficiência cardíaca, ataque cardíaco e doenças inflamatórias. A oxidação ocorre sob uma série de circunstâncias:
as células usam glicose para produzir energia
o sistema imunológico está lutando contra bactérias e criando inflamação
os corpos desintoxicam poluentes, pesticidas e fumaça de cigarro
Existem milhões de processos que ocorrem em nossos corpos em qualquer momento que pode resultar em oxidação. Aqui estão alguns sintomas:
Fadiga
Perda de memória e neblina cerebral
Dor muscular e articular
Rugas juntamente com cabelos grisalhos
Visão diminuída
Dores de cabeça e sensibilidade ao ruído
Susceptibilidade a infecções
Escolher alimentos orgânicos e evitar toxinas em seu ambiente faz uma grande diferença. Isso, junto com a redução do estresse, pode ser benéfico na redução da oxidação.
Os oxidantes são geralmente produzidos de maneira controlada, a fim de regular os processos essenciais no corpo humano, incluindo a divisão celular, inflamação, função imunológica, autofagia e resposta ao estresse. Entretanto, a produção descontrolada desses oxidantes pode contribuir para estresse oxidativo, que pode afetar a função celular, levando ao desenvolvimento de toxicidade, doença crônica e câncer. Os mecanismos antioxidantes protetores do corpo humano são regulados por uma série de vias vitais que controlam a resposta da célula aos oxidantes. O fator nuclear relacionado ao fator eritroide 2, também conhecido como Nrf2, é um regulador emergente da resistência celular a oxidantes. O objetivo do artigo abaixo é discutir e demonstrar o papel emergente do Nrf2 na função mitocondrial.
Sumário
O fator de transcrição NF-E2 p45-related factor 2 (Nrf2; nome do gene NFE2L2) permite adaptação e sobrevivência sob condições de estresse regulando a expressão gênica de diversas redes de proteínas citoprotetoras, incluindo antioxidantes, antiinflamatórios e enzimas de desintoxicação como proteínas que auxiliam no reparo ou remoção de macromoléculas danificadas. Nrf2 tem um papel crucial na manutenção da homeostase redox celular, regulando a biossíntese, utilização e regeneração de glutationa, tiorredoxina e NADPH e controlando a produção de espécies reativas de oxigênio pela mitocôndria e NADPH oxidase. Em condições homeostáticas, o Nrf2 afeta o potencial de membrana mitocondrial, a oxidação de ácidos graxos, a disponibilidade de substratos (NADH e FADH2 / succinato) para a respiração e a síntese de ATP. Sob condições de estresse ou estimulação do fator de crescimento, a ativação de Nrf2 neutraliza o aumento da produção de espécies reativas de oxigênio na mitocôndria via supra-regulação transcricional da proteína desacopladora 3 e influencia a biogênese mitocondrial ao manter os níveis de fator respiratório nuclear 1 e receptor ativado por proliferador de peroxissoma? coativador 1 ?, bem como pela promoção da biossíntese de nucleotídeos de purina. Os ativadores Nrf2 farmacológicos, como o isotiocianato sulforafano de ocorrência natural, inibem a abertura mediada por oxidante do poro de transição da permeabilidade mitocondrial e o inchaço mitocondrial. Curiosamente, descobriu-se que um composto sintético de 1,4-difenil-1,2,3-triazol, originalmente projetado como um ativador Nrf2, promove a mitofagia, contribuindo assim para a homeostase mitocondrial geral. Assim, Nrf2 é um jogador proeminente no apoio à integridade estrutural e funcional das mitocôndrias, e este papel é particularmente crucial em condições de estresse.
O Nrf2 tem um papel crucial na manutenção da homeostase redox celular.
Nrf2 afeta o potencial de membrana mitocondrial e a síntese de ATP.
Nrf2 influencia a oxidação do ácido graxo mitocondrial.
O Nrf2 suporta a integridade estrutural e funcional das mitocôndrias.
Os ativadores Nrf2 têm efeitos benéficos quando a função mitocondrial é comprometida.
Introdução
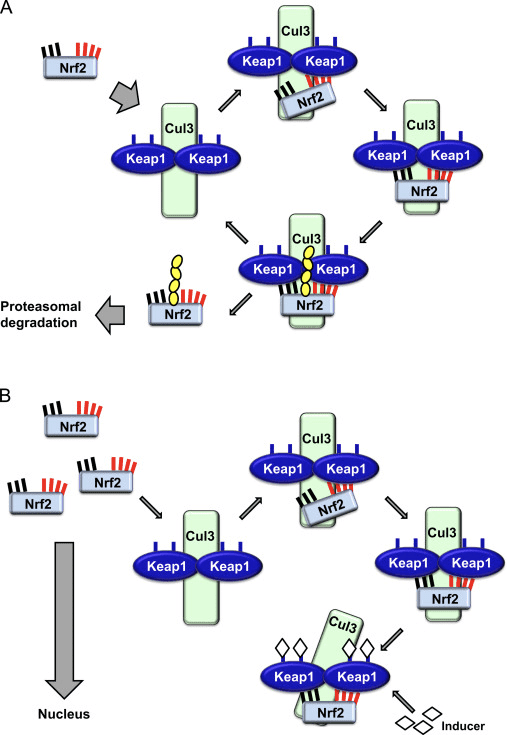
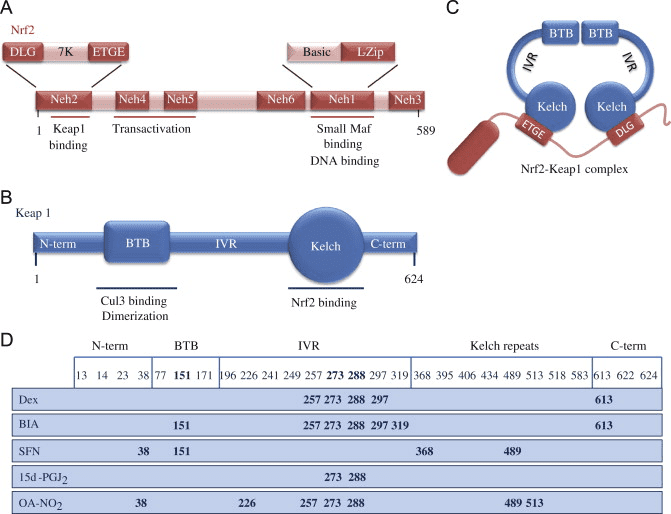
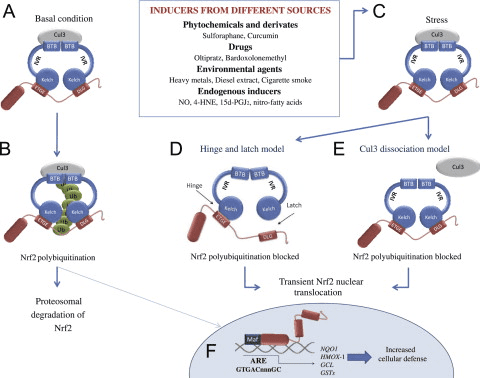
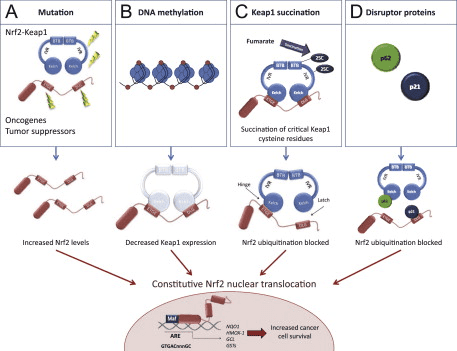
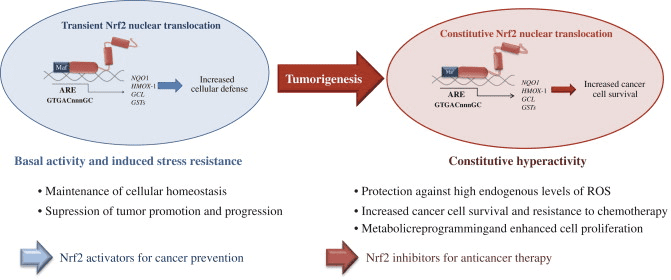
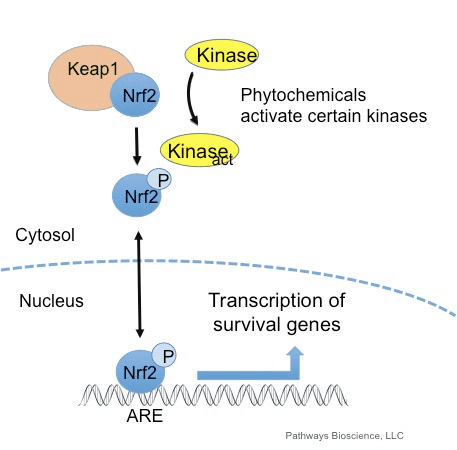
O fator de transcrição NF-E2 p45-related factor 2 (Nrf2; nome do gene NFE2L2) regula a expressão de redes de genes que codificam proteínas com diversas atividades citoprotetoras. O próprio Nrf2 é controlado principalmente no nível de estabilidade da proteína. Em condições basais, Nrf2 é uma proteína de vida curta que está sujeita a ubiquitinação contínua e degradação proteassomal. Existem três sistemas conhecidos de ubiquitina ligase que contribuem para a degradação de Nrf2. Historicamente, o primeiro regulador negativo de Nrf2 a ser descoberto foi a proteína 1 associada a ECH semelhante a Kelch (Keap1) [1], uma proteína adaptadora de substrato para Cullin 3 (Cul3) / Rbx1 ubiquitina ligase [2], [3], [ 4]. Keap1 usa um mecanismo cíclico altamente eficiente para direcionar Nrf2 para ubiquitinação e degradação proteassomal, durante o qual Keap1 é continuamente regenerado, permitindo que o ciclo prossiga (Fig. 1A) [5]. Nrf2 também está sujeito a degradação mediada por glicogênio sintase quinase (GSK) 3 /? - ubiquitina ligase baseada em Cul1 dependente de TrCP [6], [7]. Mais recentemente, foi relatado que, durante condições de estresse do retículo endoplasmático, o Nrf2 é ubiquitinado e degradado em um processo mediado pela ubiquitina ligase E3 Hrd1 [8].
Figura 1 O modelo de ligação e regeneração sequencial cíclico para a degradação mediada por Keap1 de Nrf2. (A) Nrf2 liga-se sequencialmente a um dímero Keap1 livre: primeiro através do seu domínio de ligação ETGE (varas vermelhas) de alta afinidade e depois através do seu domínio de ligação DLG (bastões negros) de baixa afinidade. Nesta conformação do complexo proteico, o Nrf2 sofre ubiquitinação e é alvo de degradação proteasomal. O Keap1 livre é regenerado e capaz de se ligar ao Nrf2 recém-convertido, e o ciclo começa novamente. (B) Os indutores (diamantes brancos) reagem com as cisteínas do sensor Keap1 (blue sticks), levando a uma mudança conformacional e prejudicando a atividade do adaptador de substrato. Keap1 livre não é regenerado, e o Nrf2 recém sintetizado se acumula e transloca para o núcleo.
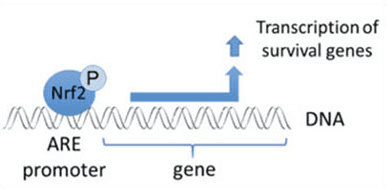
Além de servir como proteína adaptadora de substrato de ligase ubiquitina, Keap1 também é o sensor para uma ampla gama de ativadores de moléculas pequenas de Nrf2 (denominados indutores) [9]. Os indutores bloqueiam o ciclo de degradação mediada por Keap1 de Nrf2 modificando quimicamente os resíduos de cisteína específicos em Keap1 [10], [11] ou interrompendo diretamente a interface de ligação Keap1: Nrf2 [12], [13]. Consequentemente, o Nrf2 não é degradado e o fator de transcrição se acumula e transloca para o núcleo (Fig. 1B), onde forma um heterodímero com uma pequena proteína Maf; liga-se a elementos de resposta antioxidante, as regiões reguladoras a montante de seus genes-alvo; e inicia a transcrição [14], [15], [16]. A bateria de alvos Nrf2 compreende proteínas com diversas funções citoprotetoras, incluindo enzimas do metabolismo xenobiótico, proteínas com funções antioxidante e antiinflamatória, e subunidades proteossômicas, além de proteínas que regulam a homeostase redox celular e participam do metabolismo intermediário.
Nrf2: um regulador mestre da homeostase redox celular
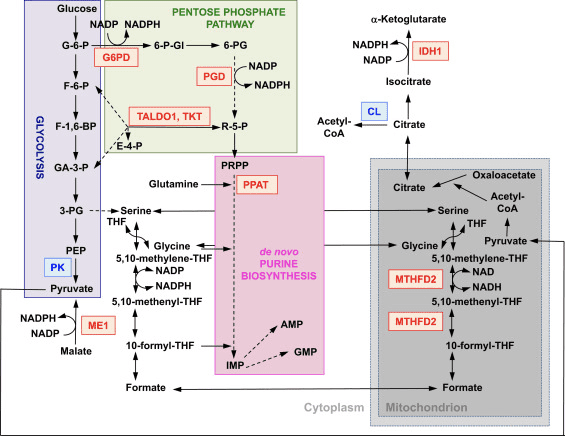
A função do Nrf2 como regulador mestre da homeostase redox celular é amplamente reconhecida. A expressão gênica das subunidades catalíticas e regulatórias da? -Glutamil cisteína ligase, a enzima que catalisa a etapa limitante da taxa na biossíntese da glutationa reduzida (GSH), é diretamente regulada pelo Nrf2 [17]. A subunidade xCT do sistema xc-, que importa cistina para as células, também é um alvo transcricional direto de Nrf2 [18]. Na célula, a cistina sofre conversão em cisteína, um precursor da biossíntese de GSH. Além de seu papel na biossíntese de GSH, o Nrf2 fornece os meios para a manutenção da glutationa em seu estado reduzido pela regulação transcricional coordenada da glutationa redutase 1 [19], [20], que reduz a glutationa oxidada a GSH usando equivalentes redutores do NADPH . O NADPH necessário é fornecido por quatro principais enzimas geradoras de NADPH, enzima málica 1 (ME1), isocitrato desidrogenase 1 (IDH1), glicose-6-fosfato desidrogenase (G6PD) e 6-fosfogluconato desidrogenase (PGD), todos os quais são regulado transcricionalmente em parte por Nrf2 (Fig. 2) [21], [22], [23], [24]. Curiosamente, o Nrf2 também regula a expressão gênica induzível das formas citosólica, microssômica e mitocondrial da aldeído desidrogenase [25], que usa o NAD (P) + como cofator, dando origem ao NAD (P) H. Na verdade, os níveis de NADPH e a razão NADPH / NADP + são mais baixos em fibroblastos embrionários isolados de camundongos Nrf2-knockout (Nrf2-KO) em comparação com as células de suas contrapartes de tipo selvagem (WT), e os níveis de NADPH diminuem após o knockdown de Nrf2 em linhas de células cancerosas com Nrf2 constitutivamente ativo [26]. Como esperado, os níveis de GSH são mais baixos nas células nas quais o Nrf2 foi interrompido; por outro lado, a ativação de Nrf2 por meios genéticos ou farmacológicos leva à regulação positiva de GSH [27], [28], [29]. É importante ressaltar que o Nrf2 também regula a expressão gênica de tioredoxina [30], [31], [32], tiorredoxina redutase 1 [28], [29], [32], [33] e sulfiredoxina [34], que são essenciais para a redução de tióis proteicos oxidados.
Figura 2 O papel do Nrf2 no metabolismo de células de proliferação rápida. Nrf2 é um regulador positivo de genes que codificam enzimas no braço oxidativo [isto é, glicose-6-fosfato desidrogenase (G6PD) e 6-fosfogluconato desidrogenase (PGD)] e no braço não oxidativo [isto é, transaldolase 1 (TALDO1) e transcetolase ( TKT)] da via da pentose fosfato. G6PD e PGD geram NADPH. O Nrf2 também regula a expressão gênica das outras duas enzimas geradoras de NADPH, a enzima málica 1 (ME1) e a isocitrato desidrogenase 1 (IDH1). A expressão gênica da fosforibosil pirofosfato amidotransferase (PPAT), que catalisa a entrada na via biossintética das purinas de novo, também é regulada positivamente pelo Nrf2, assim como a expressão da metilenotetraidrofolato desidrogenase 2 (MTHFD2), uma enzima mitocondrial com um papel crítico na fornecimento de unidades de um carbono para biossíntese de novo de purina. A piruvato quinase (PK) é negativamente regulada pelo Nrf2 e deve favorecer o acúmulo de intermediários glicolíticos e, junto com G6PD, a canalização de metabólitos através da via da pentose fosfato e a síntese de ácidos nucléicos, aminoácidos e fosfolipídios. Nrf2 regula negativamente a expressão gênica de ATP-citrato liase (CL), o que pode aumentar a disponibilidade de citrato para utilização mitocondrial ou (por meio de isocitrato) para IDH1. Vermelho e azul indicam regulação positiva e negativa, respectivamente. A mitocôndria é mostrada em cinza. Abreviações de metabólitos: G-6-P, glicose 6-fosfato; F-6-P, frutose 6-fosfato; F-1,6-BP, frutose 1,6-bisfosfato; GA-3-P, gliceraldeído 3-fosfato; 3-PG, 3-fosfoglicerato; PEP, fosfoenolpiruvato; 6-P-Gl, 6-fosfogluconolactona; 6-PG, 6-fosfogluconato; R-5-P, ribulose 5-fosfato; PRPP, 5-fosforibosil -? - 1-pirofosfato; THF, tetrahidrofolato; IMP, monofosfato de inosina; AMP, monofosfato de adenosina; GMP, monofosfato de guanosina.
Dado o papel crucial do Nrf2 como um regulador mestre da homeostase redox celular, não é surpreendente que, em comparação com as células WT, os níveis de espécies reativas de oxigênio (ROS) sejam maiores nas células em que o Nrf2 foi rompido (Nrf2-KO) [35] Essa diferença é particularmente notável após o desafio com agentes causadores de estresse oxidativo. Além disso, as células deficientes em Nrf2 são muito mais sensíveis à toxicidade de oxidantes de vários tipos e não podem ser protegidas por indutores Nrf2, que, nas mesmas condições, fornecem proteção eficiente e duradoura às células WT [29], [36] [37] Além da homeostase redox celular, o Nrf2 também é crítico para a manutenção da homeostase redox mitocondrial. Assim, comparado com WT, o total de NADH mitocondrial é significativamente aumentado em Keap1-KO e dramaticamente diminuído em células Nrf2-KO [35].
Usando imagens de células vivas, monitoramos recentemente as taxas de produção de ROS em coculturas glioneuronais primárias e fatias de tecido cerebral isoladas de camundongos WT, Nrf2-KO ou Keap1-knockdown (Keap1-KD) [38]. Como esperado, a taxa de produção de ROS foi mais rápida nas células Nrf2-KO e nos tecidos em comparação com os seus equivalentes WT. No entanto, fizemos a observação inesperada de que, em comparação com o WT, as células Keap1-KD também apresentam taxas mais altas de produção de ROS, embora a magnitude da diferença entre os genótipos WT e Keap1-KD seja menor do que entre WT e Nrf2-KO. . Em seguida, analisamos os níveis de mRNA de NOX2 e NOX4, as subunidades catalíticas das duas isoformas de NADPH oxidase (NOX) que foram implicadas em patologia cerebral, e descobrimos que NOX2 é dramaticamente aumentado sob condições de deficiência de Nrf2, enquanto NOX4 é regulado para cima quando Nrf2 é constitutivamente ativado, embora em menor grau. Quantitativamente, a magnitude da regulação positiva em células e tecidos dos camundongos mutantes se iguala ao aumento correspondente na produção de ROS [38]. Curiosamente, o Nrf2 não apenas regula a NADPH oxidase, mas as ROS produzidas pela NADPH oxidase podem ativar o Nrf2, como mostrado nas células epiteliais pulmonares e nos cardiomiócitos [39], [40]. Além disso, um estudo muito recente demonstrou que a ativação dependente de NADPH oxidase de Nrf2 constitui um importante mecanismo endógeno para proteção contra dano mitocondrial e morte celular no coração durante sobrecarga crônica de pressão [41].
Além da atividade catalítica da NADPH oxidase, a respiração mitocondrial é outra importante fonte intracelular de ROS.Ao usar a sonda específica para mitocôndrias MitoSOX, examinamos a contribuição de ROS de origem mitocondrial para a produção total de EROs em coculturas glioneuronais primárias isoladas de murganhos WT, Nrf2-KO ou Keap1-KD [38]. Como esperado, as células Nrf2-KO tiveram maiores taxas de produção de ROS mitocondrial do que WT. De acordo com os resultados para a produção total de ROS, as taxas de produção de ROS mitocondrial em Keap1-KD também foram maiores em comparação com as células WT. Importante, o bloqueio do complexo I com rotenona causou um aumento dramático na produção de ROS mitocondrial em ambas as células WT e Keap1-KD, mas não teve efeito nas células Nrf2-KO. Em contraste com o aumento esperado na produção de ROS mitocondrial em células WT após a adição de piruvato (para aumentar a disponibilidade de NADH, aumentar o potencial de membrana mitocondrial e normalizar a respiração), a produção de ROS diminuiu nas células Nrf2-KO. Juntos, esses achados sugerem fortemente que, na ausência de Nrf2: (i) a atividade do complexo I é prejudicada, (ii) a atividade prejudicada do complexo I é devido à limitação de substratos, e (iii) a atividade prejudicada do complexo Eu sou uma das principais razões para o aumento da produção de ROS mitocondrial, possivelmente devido à reversão do fluxo de elétrons do complexo II.
Nrf2 Afeta o Potencial da Membrana Mitocondrial e a Respiração
O potencial de membrana mitocondrial (?? m) é um indicador universal da saúde mitocondrial e do estado metabólico da célula. Em uma célula saudável, ?? m é mantido pela cadeia respiratória mitocondrial. Curiosamente, uma marcação isotópica estável com aminoácidos em estudo proteômico baseado em cultura na linha celular epitelial de mama humana não tumorigênica negativa para receptor de estrogênio MCF10A demonstrou que o componente da cadeia de transporte de elétrons mitocondrial NDUFA4 é regulado positivamente por ativação farmacológica (por sulforafano) de Nrf2, ao passo que a suprarregulação genética de Nrf2 (por knockdown de Keap1) leva à redução das subunidades COX2 e COX4I1 do citocromo c oxidase [42]. Um estudo do proteoma hepático usando eletroforese em gel bidimensional e espectrometria de massa de dessorção / ionização a laser assistida por matriz descobriu que Nrf2 regula a expressão da subunidade de ATP sintase? [43]. Além disso, foi relatado que a proteína mitocondrial DJ-1, que desempenha um papel na manutenção da atividade do complexo I [44], estabiliza Nrf2 [45], [46], embora os efeitos neuroprotetores da ativação farmacológica ou genética de Nrf2 são independentes de DJ-1 [47]. No entanto, as consequências dessas observações para a função mitocondrial não foram investigadas.
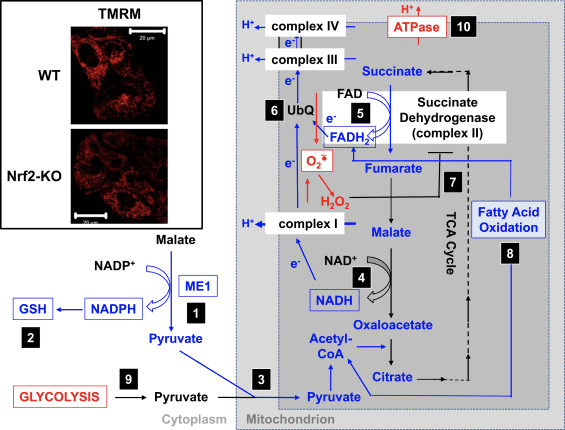
De acordo com a atividade prejudicada do complexo I sob condições de deficiência de Nrf2, o ?? m basal é menor em fibroblastos embrionários de camundongo Nrf2-KO (MEFs) e células glioneuronais primárias cultivadas em comparação com suas contrapartes WT (Fig. 3, detalhe) [35]. Em contraste, o ?? m basal é maior quando Nrf2 é regulado geneticamente constitutivamente (por knockdown ou knockout de Keap1). Essas diferenças em ?? m entre os genótipos indicam que a respiração é afetada pela atividade do Nrf2. Na verdade, a avaliação do consumo de oxigênio no estado basal revelou que, em comparação com WT, o consumo de oxigênio é menor em Nrf2-KO e Keap1-KO MEFs, em ~ 50 e ~ 35%, respectivamente.
Figura 3 Mecanismo proposto para comprometimento da função mitocondrial sob condições de deficiência de Nrf2. (1) Os níveis diminuídos de ME1, IDH1, G6PD e PGD resultam em níveis mais baixos de NADPH. (2) Os níveis de GSH também são baixos. (3) A baixa atividade de ME1 pode diminuir o pool de piruvato que entra na mitocôndria. (4) A geração de NADH é mais lenta, levando à atividade prejudicada do complexo I e ao aumento da produção de ROS mitocondrial. (5) A redução de FAD para FADH2 nas proteínas mitocondriais também é diminuída, diminuindo o fluxo de elétrons de FADH2 para UbQ e para o complexo III. (6) A formação mais lenta de UbQH2 pode diminuir a atividade enzimática da succinato desidrogenase. (7) Os níveis aumentados de ROS podem inibir ainda mais a atividade do complexo II. (8) A menor eficiência da oxidação de ácidos graxos contribui para a diminuição da disponibilidade de substrato para a respiração mitocondrial. (9) A glicólise é potencializada como mecanismo compensatório para a diminuição da produção de ATP na fosforilação oxidativa. (10) A ATP sintase opera em reverso para manter ?? m. Vermelho e azul indicam regulação positiva e regulação negativa, respectivamente. As caixas significam disponibilidade de evidências experimentais. A inserção mostra imagens de mitocôndrias de astrócitos corticais WT e Nrf2-KO visualizadas pela sonda fluorescente potenciométrica de éster metílico de tetrametilrodamina (TMRM; 25 nM). Barra de escala, 20 m.
Essas diferenças em ?? m e respiração entre os genótipos são refletidas pela taxa de utilização de substratos para a respiração mitocondrial. A aplicação de substratos para o ciclo do ácido tricarboxílico (TCA) (malato / piruvato, que por sua vez aumenta a produção do substrato do complexo I NADH) ou succinato de metila, um substrato para o complexo II, causa um aumento gradual em ?? m em ambos WT e neurônios Keap1-KD, mas a taxa de aumento é maior nas células Keap1-KD. Mais importante ainda, as formas da resposta a esses substratos do ciclo de TCA são diferentes entre os dois genótipos, pelo que o rápido aumento em ?? m nas células Keap1-KD após a adição do substrato é seguido por uma queda rápida em vez de um platô, sugerindo um aumento incomum consumo rápido de substrato. Esses achados estão de acordo com os níveis muito mais baixos (em 50-70%) de malato, piruvato e succinato que foram observados após um pulso de 1 h de glicose [U-13C6] em Keap1-KO em comparação com WT MEF células [24]. Em neurônios Nrf2-KO, apenas o piruvato é capaz de aumentar o ?? m, enquanto o malato e o succinato de metila causam despolarização leve. O efeito do Nrf2 na produção de substrato mitocondrial parece ser o principal mecanismo pelo qual o Nrf2 afeta a função mitocondrial. O índice redox NADH mitocondrial (o equilíbrio entre o consumo de NADH pelo complexo I e a produção de NADPH no ciclo de TCA) é significativamente menor nas células Nrf2-KO em comparação com suas contrapartes WT e, além disso, as taxas de regeneração dos pools de NADH e FADH2 após a inibição do complexo IV (pelo uso de NaCN) são mais lentos nas células mutantes.
Em mitocôndrias isoladas de cérebro e fígado murinos, a suplementação de substratos para o complexo I ou para o complexo II aumenta a taxa de consumo de oxigênio mais fortemente quando o Nrf2 é ativado e menos eficiente quando o Nrf2 é interrompido [35]. Assim, o malato induz uma maior taxa de consumo de oxigênio em Keap1-KD em comparação com WT, mas seu efeito é mais fraco nas mitocôndrias Nrf2-KO. Da mesma forma, na presença de rotenona (quando o complexo I é inibido), o succinato ativa o consumo de oxigênio em maior extensão em Keap1-KD em comparação com WT, enquanto a resposta em mitocôndrias Nrf2-KO é diminuída. Além disso, as culturas neuronais primárias de Nrf2-KO e camundongos são mais sensíveis à toxicidade dos inibidores do complexo II ácido 3-nitropropiônico e malonato, enquanto o transplante intraestriatal de astrócitos que superexpressam Nrf2 é protetor [48], [49]. Da mesma forma, os camundongos Nrf2-KO são mais sensíveis, enquanto a ativação genética ou farmacológica de Nrf2 tem efeitos protetores contra a neurotoxicidade causada pelo inibidor do complexo I do íon 1-metil-4-fenilpiridínio no 1-metil-4-fenil-1,2,3,6, Modelo animal 49-tetra-hidropiridina da doença de Parkinson [50], [51], [52], [53], [54], [55], [56], [57], [58], [59], [60], [61], [XNUMX].
A proporção de controle respiratório (RCR), a proporção do estado 3 (estimulado por ADP) para a respiração do estado 4 (sem ADP presente), é diminuída na ausência de Nrf2, mas o RCR é semelhante entre Keap1-KD e mitocôndrias WT [35 ] Como o RCR é uma indicação do grau de acoplamento da atividade da cadeia respiratória mitocondrial à fosforilação oxidativa, esse achado indica que a maior taxa de respiração nas mitocôndrias Keap1-KD não é devido ao desacoplamento da fosforilação oxidativa. Sugere ainda que a fosforilação oxidativa é mais eficiente quando o Nrf2 é ativado. A maior taxa de respiração nas mitocôndrias Keap1-KD é consistente com os níveis mais altos de produção de ROS mitocondrial [38], pois taxas de respiração mais altas podem levar a um aumento do vazamento de elétrons. No entanto, sob condições de estresse oxidativo, o aumento da produção de ROS é contrabalançado pela suprarregulação transcricional dependente de Nrf2 da proteína desacopladora 3 (UCP3), que aumenta a condutância de prótons da membrana interna mitocondrial e, consequentemente, diminui a produção de superóxido [62]. Muito recentemente, foi demonstrado que o produto da peroxidação lipídica 4-hidroxi-2-nonenal medeia a suprarregulação dependente de Nrf2 de UCP3 em cardiomiócitos; isso pode ser particularmente importante para proteção sob condições de estresse oxidativo, como durante a isquemiareperfusão [63].
Nrf2 Afeta a Eficiência da Fosforilação Oxidativa e a Síntese do ATP
De acordo com o efeito do Nrf2 na respiração, nas mitocôndrias do cérebro e do fígado, a deficiência de Nrf2 resulta em uma diminuição da eficiência da fosforilação oxidativa (conforme estimado pela proporção de ADP para oxigênio, que é consumido para a síntese de ATP), enquanto a ativação de Nrf2 (Keap1 -KD) tem o efeito oposto [35]. Comparado ao WT, os níveis de ATP são significativamente mais altos em células com suprarregulação constitutiva de Nrf2 e mais baixos quando Nrf2 é derrubado [64] ou interrompido [35]. Além disso, o uso de inibidores da fosforilação oxidativa (oligomicina) ou da glicólise (ácido iodoacético) revelou que o Nrf2 altera a forma como as células produzem ATP. Assim, em neurônios WT, a oligomicina causa uma queda completa no ATP e o ácido iodoacético não tem mais efeito. Notavelmente, em células Nrf2-KO, a oligomicina aumenta os níveis de ATP, que são lentamente, mas completamente, esgotados pelo ácido iodoacético, indicando que na ausência de Nrf2, a glicólise, e não a fosforilação oxidativa, é a principal fonte de produção de ATP. Curiosamente, apesar do aumento da eficiência da fosforilação oxidativa em células Keap1-KD, a adição de oligomicina resulta em uma diminuição de ~ 80% nos níveis de ATP e o ácido iodoacético causa uma redução adicional de ~ 20%. Assim, a deficiência de Nrf2 ou sua ativação constitutiva reduz a contribuição da fosforilação oxidativa e aumenta a contribuição da glicólise para a síntese de ATP. Este efeito é particularmente pronunciado quando Nrf2 está ausente e é consistente com a dependência do ?? m da presença de glicose no meio [35] e os níveis aumentados de intermediários glicolíticos (G-6-P, F-6-P , fosfato de dihidroxiacetona, piruvato e lactato) após knockdown de Nrf2 [24].
O aumento dos níveis de ATP após a inibição da F1F0-ATPase pela oligomicina indica que na ausência de Nrf2, a F1F0-ATPase funciona como ATPase e não como ATP sintase, ou seja, opera ao contrário. Essa reversão na atividade provavelmente reflete a necessidade de bombear prótons através da membrana mitocondrial interna na tentativa de manter o ?? m, o que é crucial para a integridade funcional desta organela. A reversão da função da F1F0-ATPase também é evidenciada pela despolarização mitocondrial observada após a administração de oligomicina a células Nrf2-KO, que está em nítido contraste com a hiperpolarização que ocorre em suas contrapartes deficientes em WT ou Keap1 [35]. No geral, parece que sob condições de deficiência de Nrf2, o ATP é produzido principalmente na glicólise, e esse ATP é então usado em parte pela F1F0-ATPase para manter o ?? m.
Nrf2 Melhora a Oxidação de Ácidos Graxos Mitocondriais
O efeito da deficiência de Nrf2 no ?? m é particularmente pronunciado quando as células são incubadas em meio sem glicose, e o ?? m é ~ 50% menor em Nrf2-KO em comparação com as células WT [35]. Em condições de privação de glicose, a oxidação de ácidos graxos mitocondriais (FAO) é o principal fornecedor de substratos para a respiração e fosforilação oxidativa, sugerindo que o Nrf2 pode afetar a FAO. Na verdade, a eficiência da FAO tanto para o ácido graxo saturado de cadeia longa (C16: 0), ácido palmítico e para o ácido hexanóico de cadeia curta (C6: 0) é maior em Keap1-KO MEFs e mitocôndrias isoladas de coração e fígado do que em WT homólogos, enquanto é menor nas células Nrf2-KO e mitocôndrias [65]. Esses efeitos também são altamente relevantes para os humanos: de fato, alterações metabólicas indicativas de uma melhor integração da FAO com a atividade do ciclo do TCA foram relatadas em estudos de intervenção humana com dietas ricas em glucorafanina, o precursor do clássico ativador Nrf2 sulforafano [ 66].
Durante a primeira etapa da FAO mitocondrial, o hidrogênio pró-R do carbono? Sai como um hidreto que reduz o cofator FAD para FADH2, que por sua vez transfere elétrons para a ubiquinona (UbQ) na cadeia respiratória, em última análise, contribuindo para a produção de ATP . Enquanto a estimulação de FAO por palmitoilcarnitina na ausência de glicose causa o aumento esperado nos níveis de ATP em células WT e Keap1-KO, com o aumento de ATP sendo mais rápido em células Keap1-KO, o tratamento idêntico não produz alterações de ATP em Nrf2-KO MEFs [65]. Este experimento demonstra que, na ausência de Nrf2, a FAO é suprimida e, além disso, implica a supressão da FAO como uma das razões para os níveis mais baixos de ATP em condições de deficiência de Nrf2 [35], [64].
Notavelmente, as células 293 T humanas nas quais Nrf2 foi silenciada têm uma expressão mais baixa de CPT1 e CPT2 [67], duas isoformas da carnitina palmitoiltransferase (CPT), a enzima limitante da velocidade na FAO mitocondrial. De acordo, os níveis de mRNA de Cpt1 são menores nos fígados de Nrf2-KO em comparação com os camundongos WT [68]. A CPT catalisa a transferência do grupo acilo de uma acil-CoA gorda de cadeia longa da coenzima A para l-carnitina e permite assim a importação de acilcarnitina do citoplasma para a mitocôndria. Embora isso não tenha sido examinado até o momento, é possível que além dos efeitos transcricionais sobre a expressão de CPT1, o Nrf2 também possa afetar a função dessa enzima, controlando os níveis de seu principal inibidor alostérico, a malonil-CoA. Isso porque, por um mecanismo que atualmente não é claro, o Nrf2 regula negativamente a expressão da estearoil-CoA dessaturase (SCD) [69] e citrato liase (CL) [69], [70]. Curiosamente, o knockout ou inibição de SCD leva ao aumento da fosforilação e ativação da proteína quinase ativada por AMP (AMPK) [71], [72], [73], e pode-se especular que, na ausência de Nrf2, os níveis de SCD aumentará, por sua vez, diminuindo a atividade da AMPK. Isso poderia ser ainda mais agravado pelos níveis reduzidos de proteína de AMPK que foram observados em fígados de camundongos Nrf2-KO [68], um achado que está em estreita concordância com os níveis aumentados de AMPK, que foram relatados em fígados de Keap1-KD ratos [74]. Uma conseqüência da diminuição da atividade de AMPK é o alívio de sua fosforilação inibitória (em Ser79) da acetil-CoA carboxilase (ACC) [75], que pode ser transcricionalmente regulada positivamente na ausência de Nrf2 porque é negativamente regulada pela ativação de Nrf2 [70 ]. A alta atividade de ACC, em combinação com a expressão de CL regulada positivamente que aumentará a produção de acetil-CoA, o substrato para ACC, pode, em última análise, aumentar os níveis do produto ACC, malonil-CoA. Os altos níveis de malonil-CoA inibem a CPT, diminuindo assim o transporte de ácidos graxos para a mitocôndria. Finalmente, Nrf2 regula positivamente a expressão de CD36 [76], uma translocase que importa ácidos graxos através do plasma e das membranas mitocondriais. Assim, um mecanismo pelo qual Nrf2 pode afetar a eficiência da FAO mitocondrial é através da regulação da importação de ácidos graxos de cadeia longa para as mitocôndrias.
Além da regulação direta da transcrição, o Nrf2 também pode alterar a eficiência da FAO mitocondrial por seus efeitos no metabolismo redox celular. Isso pode ser especialmente relevante quando a atividade Nrf2 é baixa ou ausente, condições que alteram o status redox celular em direção ao estado oxidado. De fato, várias enzimas da FAO foram identificadas como sensíveis a alterações redox. Uma dessas enzimas é a acil-CoA desidrogenase de cadeia muito longa (VLCAD), que contribui com mais de 80% para a atividade de desidrogenação da palmitoil-CoA em tecidos humanos [77]. Curiosamente, Hurd et al. [78] mostraram que o VLCAD contém resíduos de cisteína que alteram significativamente seu estado redox quando da exposição de mitocôndrias isoladas de coração de rato a H2O2. Além disso, a S-nitrosilação da VLCAD hepática de murino na Cys238 melhora a eficiência catalítica da enzima [79], e é provável que a oxidação da mesma cisteína possa ter o efeito oposto, diminuindo a eficiência da FAO mitocondrial. Portanto, é possível que, embora os níveis de expressão de VLCAD não sejam significativamente diferentes em MEFs de WT, Nrf2-KO ou Keap1-KO [65], a atividade enzimática de VLCAD poderia ser menor na ausência de Nrf2 devido aos níveis mais altos de ROS.
Com base em todos esses achados, pode ser proposto que (Fig. 3): na ausência de Nrf2, os níveis de NADPH são mais baixos devido à diminuição da expressão de ME1, IDH1, G6PD e PGD. Os níveis de glutationa reduzida também são menores devido à diminuição da expressão de enzimas que participam de sua biossíntese e regeneração e aos níveis mais baixos de NADPH que são necessários para a conversão da forma oxidada na forma reduzida de glutationa. A baixa expressão de ME1 diminuirá o pool de piruvato que entra na mitocôndria, com a glicólise se tornando a principal fonte de piruvato. A geração de NADH é mais lenta, levando à atividade prejudicada do complexo I e ao aumento da produção de ROS mitocondrial. A redução de FAD para FADH2 também é mais lenta, pelo menos em parte devido à oxidação de ácidos graxos menos eficiente, comprometendo o fluxo de elétrons de FADH2 para UbQ e para o complexo III. Como UbQH2 é um ativador da succinato desidrogenase [80], retardar sua formação pode diminuir a atividade enzimática da succinato desidrogenase. Os níveis aumentados de superóxido e peróxido de hidrogênio podem inibir ainda mais a atividade do complexo II [81]. A menor eficiência da oxidação de ácidos graxos contribui para a diminuição da disponibilidade de substrato para a respiração mitocondrial e produção de ATP na fosforilação oxidativa. Como mecanismo compensatório, a glicólise é aumentada. A ATP sintase funciona ao contrário, como uma ATPase, na tentativa de manter o ?? m.
Nrf2 e Biogénese Mitocondrial
Foi relatado que, em comparação com WT, os fígados de camundongos Nrf2-KO têm um conteúdo mitocondrial inferior (conforme determinado pela proporção de DNA mitocondrial para nuclear); isto é ainda diminuído por um jejum de 24 h em ambos os camundongos WT e Nrf2-KO; em contraste, embora não seja diferente do WT em condições normais de alimentação, o conteúdo mitocondrial em camundongos com alta atividade de Nrf2 não é afetado pelo jejum [82]. Curiosamente, a suplementação com o ativador Nrf2 (R) -? - ácido lipóico [83], [84], [85] promove a biogênese mitocondrial em adipócitos 3T3-L1 [86]. Duas classes de reguladores transcricionais nucleares desempenham papéis críticos na biogênese mitocondrial. A primeira classe são fatores de transcrição, como fatores respiratórios nucleares11 e 2, que controlam a expressão de genes que codificam subunidades dos cinco complexos respiratórios, componentes translacionais mitocondriais e enzimas biossintéticas heme que estão localizadas na matriz mitocondrial [88]. Piantadosi et al. [89] demonstraram que a suprarregulação transcricional dependente de Nrf2 do fator respiratório nuclear 1 promove a biogênese mitocondrial e protege contra a citotoxicidade do agente quimioterápico antraciclina cardiotóxico doxorrubicina. Em contraste, Zhang et al. [82] relataram que a ativação genética de Nrf2 não afeta a expressão de mRNA basal do fator respiratório nuclear 1 no fígado murino.
A segunda classe de reguladores transcricionais nucleares com funções críticas na biogênese mitocondrial são os coativadores transcricionais, como o receptor ativado por proliferador de peroxissoma? coativadores (PGC) 1? e 1 ?, que interagem com fatores de transcrição, a maquinaria basal de transcrição e de splicing de RNA e enzimas modificadoras de histonas [88], [90], [91]. A expressão da família de coativadores PGC1 é influenciada por vários sinais ambientais. O tratamento de fibroblastos humanos com o ativador Nrf2 sulforafano causa aumento da massa mitocondrial e indução de PGC1? e PGC1? [92], embora a dependência potencial de Nrf2 não tenha sido examinada neste estudo. No entanto, camundongos diabéticos nos quais Nrf2 é ativado pelo knockdown hipomórfico do gene Keap1 (db / db: Keap1flox / ?: Nrf2 + / +) ou interrompido (db / db: Keap1flox / ?: Nrf2? /?) Têm PGC1 hepático inferior? níveis de expressão do que os animais de controle (db / db: Keap1flox / +: Nrf2 + / +) [93]. Nenhuma diferença nos níveis de mRNA para PGC1? são vistos em fígados de camundongos não diabéticos que são WT ou Nrf2-KO, enquanto esses níveis são mais baixos em animais com superexpressão de Nrf2 (Keap1-KD e Keap1-KO específico do fígado) [82]. Notavelmente, um jejum de 24 horas aumenta os níveis de PGC1? mRNA em fígados de camundongos de todos os genótipos, mas o aumento é significativamente maior em fígados de Nrf2-KO em comparação com WT ou camundongos com superexpressão de Nrf2. Em comparação com WT, os camundongos Nrf2-KO experimentando infecção séptica ou lesão pulmonar aguda devido à infecção mostram a suprarregulação transcricional atenuada do fator respiratório nuclear 1 e PGC1? [94], [95]. Juntas, essas observações sugerem que o papel do Nrf2 na manutenção dos níveis do fator respiratório nuclear 1 e do PGC1? é complexo e se torna mais proeminente em condições de estresse.
Além da expressão de genes que codificam proteínas mitocondriais, a biogênese mitocondrial requer a síntese de nucleotídeos. A ativação genética de Nrf2 aumenta a biossíntese de purinas por meio da regulação positiva da via da pentose fosfato e do metabolismo do folato e da glutamina, particularmente em células de proliferação rápida (Fig. 2) [24]. A análise do transcriptoma de Drosophila mutante deficiente para a proteína quinase serina / treonina mitocondrial induzida por PTEN quinase 1 putativa (PINK1) mostrou que a disfunção mitocondrial leva à suprarregulação da transcrição de genes que afetam o metabolismo de nucleotídeos [96], sugerindo que a biossíntese de nucleotídeos aumentada representa um mecanismo de proteção contra as consequências neurotóxicas da deficiência de PINK1. Nrf2 regula a expressão de fosforibosil pirofosfato amidotransferase (PPAT), que catalisa a entrada na via biossintética do nucleotídeo purina de novo e a metilenotetraidrofolato desidrogenase 2 mitocondrial (MTHFD2) (Fig. 2). A última é uma enzima bifuncional com atividades de desidrogenase e ciclo-hidrolase que é crítica no fornecimento de glicina e formato como fontes de unidades de um carbono para a biossíntese de purina em células de crescimento rápido [97]. Portanto, é provável que a ativação do Nrf2 possa ser protetora e pode reverter a disfunção mitocondrial na deficiência de PINK1. De fato, a ativação farmacológica de Nrf2 por sulforafano, ou o triterpenóide RTA-408, restaura ?? me protege as células deficientes em PINK1 contra a toxicidade da dopamina [98]. Embora os mecanismos subjacentes pareçam ser complexos, juntos, esses achados indicam que a atividade do Nrf2 pode afetar a biogênese mitocondrial influenciando os níveis de expressão de fatores de transcrição críticos e coativadores, bem como aumentando a biossíntese de nucleotídeos.
Nrf2 e Integridade Mitocondrial
Embora a evidência direta nem sempre esteja disponível, existem fortes indícios de que o Nrf2 é importante para a integridade mitocondrial, particularmente sob condições de estresse oxidativo. As mitocôndrias isoladas do cérebro e fígado de ratos que receberam uma dose única do sulforafano do ativador Nrf2 são resistentes à abertura do poro de transição de permeabilidade mitocondrial (mPTP) causado pelo oxidante terc-butil-hidroperóxido [99], [100]. O mPTP, um complexo que permite que a membrana interna mitocondrial se torne permeável a moléculas com massas até 1500 Da, foi recentemente identificado como sendo formado a partir de dímeros da sintase F0F1-ATP [101]. A resistência mediada pelo sulforafano à abertura do mPTP está correlacionada com o aumento das defesas antioxidantes, e os níveis de GSH mitocondrial, glutationa peroxidase 1, enzima málica 3 e tiorredoxina 2 são todos regulados em frações mitocondriais isoladas de animais tratados com sulforafane [100].
O dano à proteína mitocondrial e o comprometimento da respiração causados pelo produto da peroxidação lipídica eletrofílica 4-hidroxi-2-nonenal são atenuados nas mitocôndrias isoladas do córtex cerebral de camundongos tratados com sulforafano [102]. Em células epiteliais renais de rato e no rim, o sulforafano é protetor contra a toxicidade induzida por cisplatina e gentamicina e perda de ?? m [103], [104]. Proteção contra um painel de oxidantes (superóxido, peróxido de hidrogênio, peroxinitrito) e eletrófilos (4-hidroxi-2-nonenal e acroleína) e um aumento nas defesas antioxidantes mitocondriais também foram observados no tratamento de células do músculo liso da aorta de rato com sulforafano [105 ] Em um modelo de lesão renal aguda induzida por contraste, o pré-condicionamento isquêmico de membro recentemente demonstrou ter efeitos protetores, incluindo inibição da abertura do mPTP e inchaço mitocondrial, pela ativação de Nrf2 conseqüente à inibição de GSK3? [106].
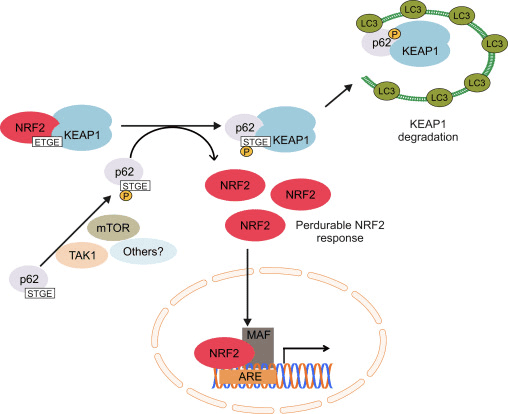
Mitofagia, o processo pelo qual mitocôndrias disfuncionais são seletivamente engolfadas por autofagossomos e entregues aos lisossomos para serem degradados e reciclados pela célula, é essencial para a homeostase mitocondrial [107], [108]. Considerando que nenhuma relação causal entre Nrf2 e mitofagia foi estabelecida, há evidências de que o fator de transcrição pode ser importante no controle de qualidade mitocondrial por desempenhar um papel na mitofagia. Isso pode ser especialmente proeminente em condições de estresse oxidativo. Assim, em um modelo de sepse, os aumentos nos níveis do marcador de autofagossomo MAP1 de cadeia leve 3-II (LC3-II) e da proteína de carga p62 em 24 h pós-infecção são suprimidos em Nrf2-KO em comparação com camundongos WT [109] . Um indutor de mitofagia de pequena molécula (denominado indutor de mitofagia mediado por p62, PMI) foi descoberto recentemente; este composto 1,4-difenil-1,2,3-triazol foi originalmente projetado como um ativador Nrf2 que interrompe a interação do fator de transcrição com Keap1 [110]. Semelhante às células em que Nrf2 é geneticamente regulado positivamente (Keap1-KD ou Keap1-KO), as células expostas a PMI têm maior ?? m em repouso. É importante ressaltar que o aumento na localização de LC3 mitocondrial que é observado após o tratamento de células WT com PMI não ocorre em células Nrf2-KO, sugerindo o envolvimento de Nrf2.
Por último, a análise ultra-estrutural das secções do fígado revelou a presença de mitocôndrias inchadas com crista reduzida e membranas rompidas nos hepatócitos de Nrf2-KO, mas não WT, ratinhos que foram alimentados com uma dieta rica em gordura durante as semanas 24; notavelmente, esses fígados mostram evidência clara de estresse oxidativo e inflamação [68]. Pode-se concluir que o Nrf2 tem um papel crítico na manutenção da integridade mitocondrial sob condições de estresse oxidativo e inflamatório.
Sulforafano e seus efeitos sobre câncer, mortalidade, envelhecimento, cérebro e comportamento, doenças cardíacas e muito mais
Os isotiocianatos são alguns dos compostos vegetais mais importantes que você pode obter em sua dieta. Neste vídeo eu faço o caso mais abrangente para eles que já foi feito. Curto período de atenção? Pule para o seu tópico favorito clicando em um dos pontos de tempo abaixo. Cronograma completo abaixo.
Seções principais:
00: 01: 14 - Câncer e mortalidade
00: 19: 04 - envelhecimento
00: 26: 30 - Cérebro e comportamento
00: 38: 06 - recapitulação final
00: 40: 27 - dose
Cronograma completo:
00: 00: 34 - Introdução do sulforafano, um dos principais focos do vídeo.
00: 01: 14 - Consumo de vegetais crucíferos e reduções na mortalidade por todas as causas.
00: 02: 12 - risco de câncer de próstata.
00: 02: 23 - risco de câncer de bexiga.
00: 02: 34 - Câncer de pulmão em risco de fumantes.
00: 02: 48 - risco de câncer de mama.
00: 03: 13 - Hipotético: e se você já tem câncer? (intervencionista)
00: 03: 35 - Mecanismo plausível que direciona os dados associativos de câncer e mortalidade.
00: 04: 38 - Sulforafano e câncer.
00: 05: 32 - Evidência animal mostrando forte efeito do extrato de brócolis no desenvolvimento do tumor de bexiga em ratos.
00: 06: 06 - Efeito da suplementação direta de sulforafano em pacientes com câncer de próstata.
00: 07: 09 - Bioacumulação de metabólitos de isotiocianato no tecido mamário atual.
00: 08: 32 - Inibição de células estaminais de cancro da mama.
00: 08: 53 - Lição de História: os brassicas foram estabelecidos como tendo propriedades de saúde mesmo na Roma antiga.
00: 09: 16 - A capacidade do Sulforaphane de aumentar a excreção de carcinógeno (benzeno, acroleína).
00: 09: 51 - NRF2 como um interruptor genético através de elementos de resposta antioxidante.
00: 10: 10 - Como a ativação de NRF2 aumenta a excreção de carcinógenos via conjugados de glutationa-S.
00: 10: 34 - As couves-de-bruxelas aumentam a glutationa-S-transferase e reduzem os danos no DNA.
00: 11: 20 - Bebida de brócolis aumenta a excreção de benzeno em 61%.
00: 13: 31 - O homogenato de brócolis aumenta as enzimas antioxidantes nas vias aéreas superiores.
00: 15: 45 - Consumo de vegetais crucíferos e mortalidade por doenças cardíacas.
00: 16: 55 - Brócolis em pó melhora os lipídios no sangue e o risco geral de doenças cardíacas em diabéticos tipo 2.
00: 19: 04 - Início da seção de envelhecimento.
00: 19: 21 - dieta enriquecida com sulforafano aumenta a vida útil de besouros de 15 a 30% (em certas condições).
00: 20: 34 - Importância da baixa inflamação para a longevidade.
00: 22: 05 - Os vegetais crucíferos e o pó de brócolis parecem reduzir uma grande variedade de marcadores inflamatórios em humanos.
00: 23: 40 - Recapitulação de vídeo intermediário: câncer, seções de envelhecimento
00: 24: 14 - Estudos com ratos sugerem que o sulforafano pode melhorar a função imunológica adaptativa na velhice.
00: 25: 18 - Sulforaphane melhorou o crescimento do cabelo em um modelo de rato de calvície. Imagem no 00: 26: 10.
00: 26: 30 - Início da seção do cérebro e comportamento.
00: 27: 18 - Efeito do extrato de brócolis no autismo.
00: 27: 48 - Efeito da glucorafanina na esquizofrenia.
00: 28: 17 - Início da discussão sobre depressão (mecanismo plausível e estudos).
00: 31: Estudo 21 - Mouse usando 10 diferentes modelos de depressão induzida por estresse mostram sulforafano igualmente eficaz como fluoxetina (prozac).
00: 32: 00 - Estudo mostra a ingestão direta de glucorafanina em camundongos é igualmente eficaz na prevenção da depressão do modelo de estresse de derrota social.
00: 33: 01 - Início da seção de neurodegeneração.
00: 33: 30 - Sulforafano e doença de Alzheimer.
00: 33: 44 - Sulforaphane e doença de Parkinson.
00: 33: 51 - Sulforaphane e doença de Hungtington.
00: 34: 13 - Sulforafano aumenta as proteínas de choque térmico.
00: 34: 43 - Início da seção de traumatismo cranioencefálico.
00: 35: 01 - Sulforafano injetado imediatamente após o TBI melhora a memória (estudo do mouse).
00: 35: 55 - Sulforafano e plasticidade neuronal.
00: 36: 32 - Sulforaphane melhora o aprendizado em modelos de diabetes tipo II em camundongos.
00: 37: 19 - Sulforafano e distrofia muscular de duchenne.
00: 37: 44 - Inibição da miostatina em células satélites musculares (in vitro).
00: 38: 06 - Recapitulação de vídeo tardio: mortalidade e câncer, danos no DNA, estresse oxidativo e inflamação, excreção de benzeno, doença cardiovascular, diabetes tipo II, efeitos no cérebro (depressão, autismo, esquizofrenia, neurodegeneração), via NRF2.
00: 40: 27 - Pensamentos em descobrir uma dose de brotos de brócolis ou sulforafano.
00: 41: 01 - Anedotas sobre brotar em casa.
00: 43: 14 - Nas temperaturas de cozimento e atividade de sulforafano.
00: 43: 45 - Conversão da bactéria intestinal do sulforafano da glucorafanina.
00: 44: 24 - Os suplementos funcionam melhor quando combinados com a mirosinase ativa de vegetais.
00: 44: 56 - Técnicas de cozinha e vegetais crucíferos.
00: 46: 06 - Isotiocianatos como sendo goitrogénios.
Nrf2 é um fator de transcrição que desempenha um papel importante no sistema de defesa antioxidante celular do corpo humano. O elemento antioxidante responsivo, ou ARE, é um mecanismo regulador dos genes. Muitos estudos de pesquisa demonstraram que o Nrf2, ou fator 2 relacionado a NF-E2, regula uma ampla variedade de genes dirigidos por ARE ao longo de vários tipos de células. Verificou-se também que o Nrf2 desempenha um papel essencial na protecção celular e anti-carcinogenicidade, o que demonstra que o Nrf2 pode ser um tratamento eficaz no tratamento de doenças neurodegenerativas e cancros que se acredita serem causados por stress oxidativo. Dr. Alex Jimenez DC, Insight CCST
Observações finais
Embora muitas questões ainda permaneçam abertas, as evidências experimentais disponíveis indicam claramente que o Nrf2 é um participante importante na manutenção da homeostase mitocondrial e integridade estrutural. Esse papel torna-se particularmente crítico sob condições de estresse oxidativo, eletrofílico e inflamatório quando a capacidade de regular positivamente as respostas citoprotetoras mediadas pelo Nrf2 influencia a saúde geral e a sobrevivência da célula e do organismo. O papel do Nrf2 na função mitocondrial representa outra camada dos mecanismos citoprotetores amplos orquestrados por este fator de transcrição. Como muitas condições patológicas humanas têm estresse oxidativo, inflamação e disfunção mitocondrial como componentes essenciais de sua patogênese, a ativação farmacológica de Nrf2 é promissora na prevenção e tratamento de doenças. A compreensão abrangente dos mecanismos precisos pelos quais o Nrf2 afeta a função mitocondrial é essencial para o planejamento racional de futuros ensaios clínicos e pode oferecer novos biomarcadores para o monitoramento da eficácia terapêutica.
O objetivo do artigo acima foi discutir - bem como demonstrar - o papel emergente do Nrf2 na função mitocondrial. Nrf2, ou fator nuclear fator eritroide 2-relacionado, é um regulador emergente da resistência celular a oxidantes que podem contribuir para o estresse oxidativo, afetando a função celular e levando ao desenvolvimento de toxicidade, doenças crônicas e até câncer. Embora a produção de oxidantes no corpo humano possa servir a vários propósitos, incluindo divisão celular, inflamação, função imunológica, autofagia e resposta ao estresse, é essencial controlar sua superprodução para prevenir problemas de saúde. O escopo de nossas informações é limitado a questões de quiropraxia e saúde da coluna vertebral. Para discutir o assunto, sinta-se à vontade para perguntar ao Dr. Jimenez ou entre em contato conosco em 915-850-0900 .
Discussão de tópico adicional: Dor nas costas aguda
Dor nas costasEssa é uma das causas mais prevalentes de deficiência e dias perdidos no trabalho em todo o mundo. A dor nas costas é atribuída ao segundo motivo mais comum para as consultas médicas, superada apenas pelas infecções respiratórias superiores. Aproximadamente 80 por cento da população sentirá dor nas costas pelo menos uma vez ao longo da vida. A coluna vertebral é uma estrutura complexa composta de ossos, articulações, ligamentos e músculos, entre outros tecidos moles. Por causa disso, lesões e / ou condições agravadas, comohérnia de discos, pode eventualmente levar a sintomas de dor nas costas. Lesões esportivas ou acidentes automobilísticos costumam ser a causa mais frequente de dores nas costas; no entanto, às vezes os movimentos mais simples podem ter resultados dolorosos. Felizmente, opções alternativas de tratamento, como tratamento quiroprático, podem ajudar a aliviar a dor nas costas por meio do uso de ajustes da coluna vertebral e manipulações manuais, melhorando, em última análise, o alívio da dor.
Nrf2 apoia a ativação de um grupo de enzimas e genes antioxidantes e desintoxicantes que protegem o corpo humano dos efeitos dos problemas de saúde associados ao aumento dos níveis de estresse oxidativo, como a doença de Alzheimer. Foi demonstrado que uma variedade de substâncias naturais ativam a via Nrf2, que pode ajudar a controlar os sintomas de doenças neurodegenerativas. O objetivo do artigo abaixo é discutir o papel central do Nrf2 causado pela inflamação crônica.
Sumário
A inflamação é a característica mais comum de muitas doenças crônicas e complicações, enquanto desempenha um papel crítico na carcinogênese. Vários estudos têm demonstrado que o Nrf2 contribui para o processo antiinflamatório orquestrando o recrutamento de células inflamatórias e regulando a expressão gênica por meio do elemento de resposta antioxidante (ERA). A via de sinalização Keap1 (proteína associada a ECH semelhante a Kelch) / Nrf2 (fator 2 relacionado a p45 NF-E2) / ARE regula principalmente a expressão de genes antiinflamatórios e inibe a progressão da inflamação. Portanto, a identificação de novos fitoquímicos antiinflamatórios dependentes de Nrf2 tornou-se um ponto chave na descoberta de medicamentos. Nesta revisão, discutimos os membros da via de sinalização Keap1 / Nrf2 / ARE e seus genes a jusante, os efeitos dessa via em modelos animais de doenças inflamatórias e o crosstalk com a via NF-? B. Além disso, também discutimos sobre a regulação do inflamassoma de NLRP3 por Nrf2. Além disso, resumimos o cenário atual de desenvolvimento de fitoquímicos antiinflamatórios e outros que medeiam a via de sinalização Nrf2 / ARE.
A inflamação é um processo complexo que ocorre quando os tecidos são infectados ou lesionados por estímulos nocivos, como patógenos, danos ou irritantes. Células imunes, vasos sangüíneos e mediadores moleculares estão envolvidos nessa resposta protetora [1]. A inflamação também é um fenômeno patológico associado a uma variedade de estados de doença induzidos principalmente por fatores físicos, químicos, biológicos e psicológicos. O objetivo da inflamação é limitar e eliminar as causas do dano celular, limpar e / ou absorver células e tecidos necróticos e iniciar o reparo tecidual. Distinguem-se duas formas distintas de inflamação: aguda e crônica. A inflamação aguda é autolimitada e benéfica para o hospedeiro, mas a inflamação crônica prolongada é uma característica comum de muitas doenças crônicas e complicações. A infiltração direta por muitas células imunes mononucleares, como monócitos, macrófagos, linfócitos e plasmócitos, bem como a produção de citocinas inflamatórias, levam à inflamação crônica. É reconhecido que a inflamação crônica desempenha um papel crítico na carcinogênese [2]. Em geral, ambas as vias de sinalização pró e anti-inflamatória interagem no processo inflamatório normal.
No processo inflamatório patológico, mastócitos, monócitos, macrófagos, linfócitos e outras células do sistema imunológico são ativados primeiro. Em seguida, as células são recrutadas para o local da lesão, resultando na geração de espécies reativas de oxigênio (ROS) que danificam macromoléculas, incluindo DNA. Ao mesmo tempo, essas células inflamatórias também produzem grandes quantidades de mediadores inflamatórios, como citocinas, quimiocinas e prostaglandinas. Esses mediadores recrutam ainda macrófagos para locais localizados de inflamação e ativam diretamente várias cascatas de transdução de sinal e fatores de transcrição associados à inflamação. As vias de sinalização NF-? B (fator nuclear kappa B), MAPK (proteína quinase ativada por mitogênio) e JAK (quinase janus) -STAT (transdutores de sinal e ativadores de transcrição) estão envolvidos no desenvolvimento da via clássica da inflamação [3], [4], [5]. Estudos anteriores revelaram que o fator de transcrição Nrf2 (fator 2 relacionado a NF-E45 p2) regula a expressão de enzimas desintoxicantes de fase II, incluindo NADPH, NAD (P) H quinona oxidorredutase 1, glutationa peroxidase, ferritina, heme oxigenase-1 (HO -1), e genes antioxidantes que protegem as células de várias lesões por meio de seus efeitos antiinflamatórios, influenciando assim o curso da doença [6], [7], [8].
Considerando esses achados notáveis, o desenvolvimento de drogas terapêuticas direcionadas para doenças inflamatórias através de vias de sinalização tem atraído muito interesse nos últimos anos. Nesta revisão, resumimos a pesquisa sobre a via de sinalização Keap1 (proteína associada à ECH tipo Kelch) / Nrf2 (fator 2 relacionado à NF-E45 p2) / ARE (elemento de resposta antioxidante) na inflamação.
Estrutura e Regulamentação da Nrf2
Regulamento Nrf1 dependente de Keap2
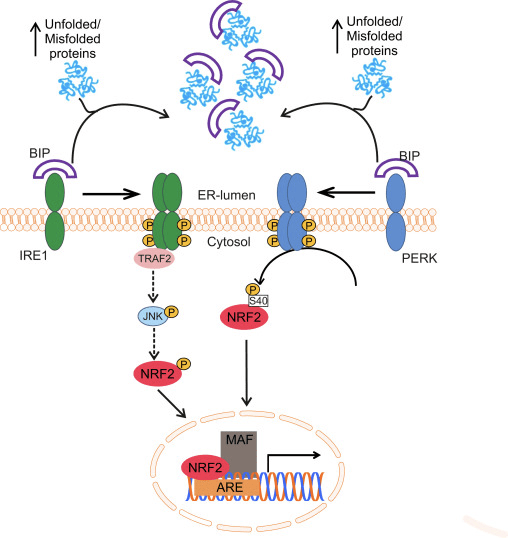
Nrf2 pertence à subfamília Cap n Collar (CNC) e compreende em sete domínios funcionais, Neh (homologia Nrf2-ECH) 1 a Neh7 [9], [10]. Neh1 é um domínio CNC-bZIP que permite que Nrf2 heterodimerize com proteína de pequeno fibrossarcoma músculoaponeurótico (Maf), DNA e outros parceiros de transcrição, bem como formando um complexo nuclear com a enzima de conjugação de ubiquitina UbcM2 [11], [12]. Neh2 contém dois motivos importantes conhecidos como DLG e ETGE, que são essenciais para a interação entre Nrf2 e seu regulador negativo Keap1 [13], [14].
Keap1 é um adaptador de substrato para a uniquitina ligase E3 baseada em culina, que inibe a atividade transcricional de Nrf2 através da ubiquitinação e degradação proteasomal sob condições normais [15], [16], [17]. Os domínios KELCH do homodímero Keap1 ligam-se aos motivos DLG e ETGE do domínio Nrf2-Neh2 no citosol, onde o ETGE atua como uma charneira com maior afinidade e o DLG atua como um trinco [18]. Sob estresse oxidativo ou por exposição a ativadores Nrf2, o Nrf2 dissocia-se da ligação Keap1 devido à modificação do tiol dos resíduos de cisteína Keap1 que, em última análise, previne a ubiquitinação Nrf2 e a degradação proteasomal [19]. Em seguida, o Nrf2 transloca-se para o núcleo, heterodimeriza-se com pequenas proteínas Maf e transactiva uma bateria ARE de genes (Fig. 1A). O terminal carboxi de Neh3 atua como um domínio de transativação interagindo com o co-ativador de transcrição conhecido como CHD6 (cromo-ATPase / proteína de ligação ao DNA da helicase) [20]. Neh4 e Neh5 também atuam como domínios de transativação, mas se ligam a outro co-ativador transcricional conhecido como CBP (proteína de ligação à proteína de ligação ao elemento de resposta cAMP) [21]. Além disso, o Neh4 e o Neh5 interagem com o cofactor nuclear RAC3 / AIB1 / SRC-3, conduzindo a uma expressão aumentada do gene ARE de Nrf2 [22]. O Neh5 possui um sinal de exportação nuclear sensível a redox que é crucial para a regulação e localização celular de Nrf2 [23].
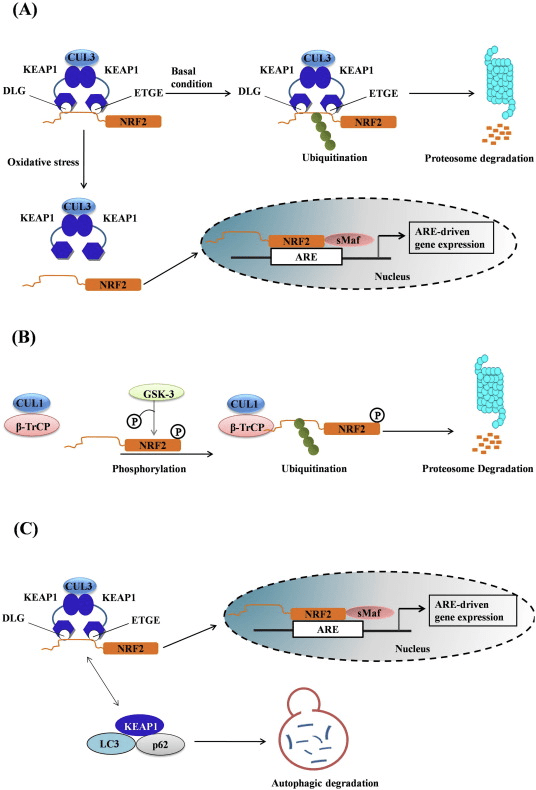
Figura 1 Keap1 dependente e independente de regulamento de Nrf2. (A) Em condições basais, Nrf2 é sequestrado com Keap1 por seus dois motivos (ETGE e DLG) que leva à ubiquitinação mediada por CUL3 seguida pela degradação do proteassoma. Sob estresse oxidativo, o Nrf2 se dissocia do Keap1, se transloca para o núcleo e ativa a bateria do gene ARE. (B) GSK3 fosforila Nrf2 e isso facilita o reconhecimento de Nrf2 por? -TrCP para ubiquitinação mediada por CUL1 e subsequente degradação do proteassoma. (C) p62 é sequestrado com Keap1, levando à sua degradação autofágica, liberação de Nrf2 e aumento da sinalização de Nrf2.
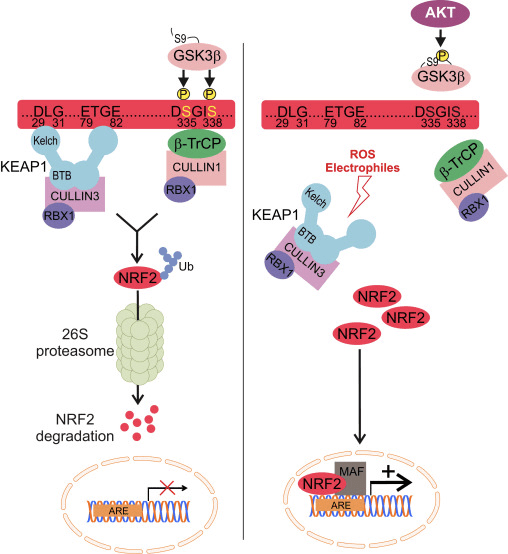
Regulamento Nrf1 independente de Keap2
Evidências emergentes revelaram um novo mecanismo de regulação do Nrf2 que é independente do Keap1. O domínio Neh6 rico em serina de Nrf2 desempenha um papel crucial nesta regulação, ligando-se com os seus dois motivos (DSGIS e DSAPGS) à proteína contendo repetição de? -Transducina (? -TrCP) [24]. ? -TrCP é um receptor de substrato para o complexo de ubiquitina ligase Skp1 Cul1 Rbx1 / Roc1 que tem como alvo Nrf2 para ubiquitinação e degradação proteassomal. A glicogênio sintase quinase-3 é uma proteína crucial envolvida na estabilização e regulação do Nrf1 independente de Keap2; ele fosforila Nrf2 no domínio Neh6 para facilitar o reconhecimento de Nrf2 por? -TrCP e subsequente degradação da proteína [25] (Fig. 1B).
Outros Reguladores Nrf2
Outra linha de evidência revelou uma via não canônica de ativação de Nrf62 dependente de p2 na qual p62 sequestra Keap1 para degradação autofágica que finalmente leva à estabilização de Nrf2 e a transativação de genes dependentes de Nrf2 [26], [27], [ 28], [29] (Fig. 1C).
O acúmulo de evidências sugere que vários miRNAs desempenham um papel importante na regulação da atividade do Nrf2 [30]. Sangokoya et al. [31] demonstraram que o miR-144 diminui diretamente a atividade do Nrf2 na linha de células de linfoblasto K562, células progenitoras eritróides humanas primárias e reticulócitos da doença falciforme. Outro estudo interessante em células epiteliais da mama humana demonstrou que o miR-28 inibe o Nrf2 por meio de um mecanismo independente de Keap1 [32]. Da mesma forma, miRNAs como miR-153, miR-27a, miR-142-5p e miR144 regulam negativamente a expressão de Nrf2 na linha celular neuronal SH-SY5Y [33]. Singh et al. [34] demonstraram que a expressão ectópica de miR-93 diminui a expressão de genes regulados por Nrf2 em um modelo de carcinogênese mamária induzida por 17? -Estradiol (E2).
Uma descoberta recente de nosso laboratório identificou um inibidor endógeno de Nrf2 conhecido como receptor alfa retinóico X (RXR?). RXR? é um receptor nuclear, interage com o domínio Neh7 de Nrf2 (resíduos de aminoácidos 209 316) por meio de seu domínio de ligação ao DNA (DBD) e inibe especificamente a atividade de Nrf2 no núcleo. Além disso, outros receptores nucleares, como receptor-? Ativado por proliferador de peroxissoma, ER ?, receptor- relacionado ao estrogênio e receptores de glicocorticóides também foram relatados como inibidores endógenos da atividade de Nrf2 [9], [10].
Papel Anti-Inflamatório do Eixo Nrf2 / HO-1
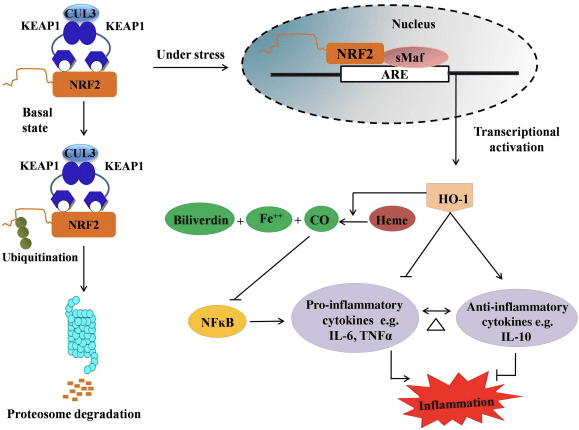
HO-1 é a isoforma indutível e a enzima limitante da taxa que catalisa a degradação do heme em monóxido de carbono (CO) e ferro livre, e biliverdina a bilirrubina. A degradação enzimática do heme livre pró-inflamatório, bem como a produção de compostos antiinflamatórios, como CO e bilirrubina, desempenham papéis importantes na manutenção dos efeitos protetores do HO-1 (Fig. 2).
Figura 2 Visão geral do caminho Nrf2 / HO-1. Em condições basais, o Nrf2 liga-se ao seu repressor Keap1, o que leva à ubiquitinação seguida pela degradação do proteassoma. Durante o estresse oxidativo, o Nrf2 livre se transloca para o núcleo, onde se dimeriza com membros da pequena família Maf e se liga a genes ARE, como HO-1. O HO-1 regulado positivamente catalisa o heme em CO, bilirrubina e ferro livre. O CO atua como um inibidor da via do NF-? B que leva à diminuição da expressão de citocinas pró-inflamatórias, enquanto a bilirrubina também atua como antioxidante. Além disso, HO-1 inibe diretamente as citocinas pró-inflamatórias, bem como ativa as citocinas antiinflamatórias, levando ao equilíbrio do processo inflamatório.
O Nrf2 induz o gene HO-1 aumentando a expressão do mRNA e da proteína e é um dos genes regulados pelo Nrf2 clássico amplamente utilizado em vários estudos in vitro e in vivo. Vários estudos demonstraram que HO-1 e seus metabólitos têm efeitos antiinflamatórios significativos mediados por Nrf2. A elevação da expressão de HO-1 que é mediada por Nrf2 ativado leva à inibição dos resultados de sinalização de NF? B na redução da lesão da mucosa intestinal e disfunção da junção apertada no modelo de transplante de fígado de rato Sprague-Dawley macho [35]. A regulação positiva da expressão de HO-2 dependente de Nrf1 pode proteger mioblastos C2C12 derivados de camundongo da citotoxicidade de H2O2 [36]. HO-2 dependente de Nrf1 tem um impacto nas respostas inflamatórias mediadas por lipopolissacarídeos (LPS) em RAW264.7- ou macrófagos de células espumosas derivadas de macrófagos peritoneais de camundongo. A atividade do Nrf2 dessensibiliza o fenótipo dos macrófagos de células espumosas e evita a inflamação imoderada dos macrófagos, que desempenham um papel importante na progressão da aterosclerose [37]. O eixo Nrf2 / HO-1 afeta células microgliais BV2 de camundongo induzidas por LPS e células HT22 de hipocampo de camundongo, com impacto na neuroinflamação. Regulação positiva da expressão de HO-1 através da via Nrf2 em células da microglia BV2 de camundongo que defendem a morte celular de células HT22 do hipocampo de camundongo [38]. Além disso, moléculas híbridas à base de cobalto (HYCOs) que combinam um indutor de Nrf2 com um liberador de monóxido de carbono (CO) aumentam a expressão de Nrf2 / HO-1, liberam CO e exercem atividade antiinflamatória in vitro. Os HYCOs também regulam positivamente a HO-1 no tecido e liberam CO no sangue após a administração in vivo, apoiando seu uso potencial contra condições inflamatórias [39]. A regulação positiva de Nrf2 / HO-1 reduz a inflamação, aumentando a atividade eferocítica de macrófagos murinos tratados com cloraminas taurinas [40]. Em conjunto, os modelos experimentais explicados acima revelaram que o eixo Nrf2 / HO-1 desempenha um papel importante na função anti-inflamatória, sugerindo que Nrf2 é um alvo terapêutico em doenças associadas à inflamação.
Além disso, os subprodutos de HO-1, como CO, bilirrubina, atuam como um poderoso antioxidante durante o estresse oxidativo e danos celulares [41], [42]; suprime encefalomielite autoimune e hepatite [43], [44]; e protege ratos e camundongos contra o choque endotóxico, impedindo a geração de iNOS e NO [45], [46], [47]. Além disso, a Bilirrubina reduz a ativação e disfunção endotelial [48]. Curiosamente, a bilirrubina reduz a transmigração de leucócitos endoteliais através da molécula de adesão 1 [49]. Estas referências específicas indicando não apenas HO-1 atua como um potente agente anti-inflamatório, mas também seus metabólitos.
Mediadores Inflamatórios e Enzimas Inibidos por Nrf2
Citocinas e quimiocinas
As citocinas são proteínas e polipeptídeos de baixo peso molecular secretados por uma variedade de células; eles regulam o crescimento celular, a diferenciação e a função imunológica, e estão envolvidos na inflamação e na cicatrização de feridas. As citocinas incluem interleucinas (ILs), interferons, fator de necrose tumoral (TNF), fator estimulador de colônias, quimiocinas e fatores de crescimento. Algumas citocinas são contadas como mediadores pró-inflamatórios, enquanto outras têm funções antiinflamatórias. A exposição ao estresse oxidativo resulta na superprodução de citocinas que causa estresse oxidativo nas células-alvo. Várias citocinas pró-inflamatórias são superproduzidas quando o NF-? B é ativado pelo estresse oxidativo. Além disso, o estresse oxidativo pró-inflamatório causa uma ativação adicional de NF-? B e a superprodução de citocinas. A ativação do sistema Nrf2 / ARE desempenha um papel importante na interrupção deste ciclo. As quimiocinas são uma família de pequenas citocinas, cujo papel principal é guiar a migração de células inflamatórias. Eles funcionam principalmente como quimioatraentes para leucócitos, monócitos, neutrófilos e outras células efetoras.
Foi relatado que a ativação de Nrf2 previne a suprarregulação da transcrição induzida por LPS de citocinas pró-inflamatórias, incluindo IL-6 e IL-1? [50]. IL-1? e a produção de IL-6 também está aumentada em Nrf2? /? camundongos com colite induzida por sulfato de dextrana [51], [52]. Nrf2 inibe a produção de IL-17 a jusante e outros fatores inflamatórios Th1 e Th17, e suprime o processo da doença em um modelo experimental de esclerose múltipla, encefalite autoimune [53]. Os genes antioxidantes dependentes de Nrf2 HO-1, NQO-1, Gclc e Gclm bloqueiam TNF- ?, IL-6, proteína quimioatrativa de monócitos-1 (MCP1), proteína inflamatória de macrófagos-2 (MIP2) e processos inflamatórios mediadores. Mas no caso de camundongos knockout para Nrf2, o efeito antiinflamatório não ocorre [54]. Neutrófilos peritoneais de camundongos knockout para Nrf2 tratados com LPS têm níveis significativamente mais elevados de citocinas (TNF- <6> e IL-1) e quimiocinas (MCP2 e MIP54) do que células do tipo selvagem (WT) [2]. In vitro, a transferência do gene Nrf1 para células do músculo liso da aorta humana e de coelho suprime a secreção de MCP8 [55], [2], e a expressão de HO-1 dependente de Nrf1 suprime TNF -? - NF-? B e MCP-56 estimulados secreção em células endoteliais da veia umbilical humana [2]. Esses achados sugerem que, em resposta a estímulos inflamatórios, a regulação positiva da sinalização de NrfXNUMX inibe a superprodução de citocinas e quimiocinas pró-inflamatórias, além de limitar a ativação de NF-? B.
Moléculas de Adesão Celular
As moléculas de adesão celular (CAMs) são proteínas que se ligam às células ou à matriz extracelular. Localizados na superfície celular, eles estão envolvidos no reconhecimento celular, ativação celular, transdução de sinal, proliferação e diferenciação. Entre as CAMs, ICAM-1 e VCAM-1 são membros importantes da superfamília das imunoglobulinas. ICAM-1 está presente em baixas concentrações em leucócitos e membranas de células endoteliais. Após a estimulação da citocina, a concentração aumenta significativamente. ICAM-1 pode ser induzido por IL-1 e TNF e é expresso pelo endotélio vascular, macrófagos e linfócitos. É um ligante da integrina, um receptor encontrado nos leucócitos. Quando a ponte ICAM-1-integrina é ativada, os leucócitos se ligam às células endoteliais e então migram para os tecidos subendoteliais [57]. O VCAM-1 medeia a adesão de linfócitos, monócitos, eosinófilos e basófilos ao endotélio vascular e contribui para o recrutamento de leucócitos, o que acaba levando a danos nos tecidos devido ao estresse oxidativo. Nrf2 inibe a atividade do promotor de VCAM-1 [58]. O gene a jusante HO-2 regulado por Nrf1 pode afetar a expressão de E-selectina e VCAM-1, moléculas de adesão associadas a células endoteliais [59]. A expressão pulmonar de vários CAMs, como CD-14, TREM1, SELE, SELP e VCAM-1 são significativamente maiores em Nrf2? /? camundongos do que em camundongos Nrf2 + / + [60]. O Nrf2 em células endoteliais da aorta humana suprime a expressão de VCAM-1 induzida por TNF -? E interfere com a adesão de células U937 monocíticas induzida por TNF -? [8]. A superexpressão de Nrf2 também inibe a expressão do gene VCAM-1 induzida por TNF -? - em células endoteliais microvasculares humanas [61]. O ácido 3-hidroxiantranílico (HA) antioxidante de ocorrência natural, um dos metabólitos de l-triptofano formado in vivo ao longo da rota metabólica conhecida como via da quinurenina durante a inflamação ou infecção, induz a expressão de HO-1 e estimula Nrf2 no umbilical humano células endoteliais da veia (HUVECs). A expressão de HO-2 dependente de Nrf1 induzida por HA inibe a secreção de MCP-1, a expressão de VCAM-1 e a ativação de NF-kB associada a lesão vascular e inflamação na aterosclerose [56]. O derivado de chalcona sintético antiproliferativo e antiinflamatório 2?, 4?, 6? -Tris (metoximetoxi) chalcona inibe ICAM-1, a citocina pró-inflamatória IL-1? E TNF-? expressão em tecido colônico de camundongos tratados com ácido trinitrobenzenossulfônico [62]. A regulação positiva de Nrf2 inibe a expressão de ICAM-1 induzida por TNF -? - em células epiteliais pigmentares da retina humanas tratadas com licopeno [63]. Todos esses estudos sugerem que o Nrf2 desempenha um papel fundamental no processo inflamatório, regulando a migração e infiltração de células inflamatórias para o tecido inflamado.
Metaloproteinases de Matriz (MMPs)
As MMPs estão amplamente presentes na matriz extracelular e estão envolvidas em processos fisiológicos e patológicos, como proliferação celular, migração, diferenciação, cicatrização de feridas, angiogênese, apoptose e metástase tumoral. Foi relatado que o eixo Nrf2 / HO-1 inibe MMP-9 em macrófagos e MMP-7 em células epiteliais intestinais humanas, e isso é benéfico no tratamento de doença inflamatória intestinal [62], [64]. O dano cutâneo induzido pela irradiação UV é mais grave em Nrf2-knockout do que em camundongos WT e o nível de MMP-9 é significativamente mais alto, indicando que Nrf2 reduz a expressão de MMP-9. Portanto, o Nrf2 é considerado protetor contra a irradiação UV [65]. Outro estudo também relatou que a ativação transcricional regulada para baixo de MMP-9 na invasão de células tumorais e inflamação é regulada através da inibição da via de sinalização de NF-kB [66]. Na lesão traumática da medula espinhal, a via de sinalização NF-kB também participa da regulação dos níveis de mRNA de MMP-9 [67]. Portanto, na inflamação, a regulação das MMPs é afetada diretamente pela via do Nrf2 ou indiretamente pela via do NF-? B influenciada pelo Nrf2.
Cicloxigenase-2 (COX2) e óxido nítrico sintético indutível (INOS)
Uma série de experimentos em camundongos knockout para Nrf2 demonstrou seu papel crucial na inflamação e na regulação de genes pró-inflamatórios como COX-2 e iNOS. Pela primeira vez, Khor et al. relataram expressão aumentada de citocinas pró-inflamatórias, como COX-2 e iNOS, nos tecidos do cólon de Nrf2? /? camundongos comparados com camundongos WT Nrf2 + / +, indicando que Nrf2 suprime sua atividade [51]. Outro relato de pré-tratamento com sulforafano, um dos conhecidos ativadores Nrf2 presentes em vegetais crucíferos, demonstrou seu efeito antiinflamatório de inibir a expressão de TNF- ?, IL-1 ?, COX-2 e iNOS em ambos os mRNA e os níveis de proteína em macrófagos peritoneais primários de camundongos Nrf2 + / + em comparação com aqueles de Nrf2? /? ratos [68]. Da mesma forma, o hipocampo de camundongos nocaute para Nrf2 com inflamação induzida por LPS também mostra maior expressão de marcadores de inflamação, como iNOS, IL-6 e TNF-? do que ratos WT [69]. Da mesma forma, camundongos knockout para Nrf2 são hipersensíveis ao estresse oxidativo induzido por 1-metil-4-fenil-1,2,3,6-tetra-hidropiridina, bem como apresentam aumento dos níveis de mRNA e proteína de marcadores de inflamação, como COX-2, iNOS , IL-6 e TNF-? [70]. Além disso, os fígados de Nrf2? /? camundongos desafiados com dieta deficiente em metionina e colina têm expressão de mRNA de Cox5 e iNOS ~ 2 vezes maior do que aqueles de camundongos WT na mesma dieta, sugerindo um papel antiinflamatório de Nrf2 [71]. Recentemente, Kim et al. demonstraram que o fitoquímico etilpiruvato exerce seus efeitos antiinflamatórios e antioxidantes por diminuir a expressão de iNOS por meio da sinalização de Nrf2 em células BV2. Eles mostraram que o piruvato de etila induz a translocação nuclear de Nrf2, o que acaba por inibir a interação entre p65 e p300, levando à diminuição da expressão de iNOS [72]. Além disso, o carbazol análogo LCY-2-CHO ativa Nrf2 e causa sua translocação nuclear, levando à supressão da expressão de COX2 e iNOS [73] em células do músculo liso vascular da aorta de rato.
Papel paradoxal de Nrf2 na regulação de NLRP3 iIflammasome Activity
A família NLR, domínio da pirina contendo 3 (NLRP3), inflamassoma é um complexo multiproteico que funciona como um receptor de reconhecimento de patógenos (PRR) e reconhece a ampla gama de sinais de estresse oxidativo microbiano, como padrões moleculares associados a patógenos (PAMPs), Danos- moléculas de padrão molecular associadas (DAMPs) e ROS [74]. O inflamassoma de NLRP3 ativado medeia a clivagem da caspase-1 e a secreção da citocina pró-inflamatória interleucina-1? (IL-1?) Que finalmente induz o processo de morte celular conhecido como piroptose, que protege os hospedeiros contra uma ampla gama de patógenos [75]. No entanto, a ativação aberrante do inflamassoma está associada a doenças de dobramento incorreto de proteínas, como encefalopatias espongiformes transmissíveis, doença de Alzheimer, doença de Parkinson e também diabetes tipo 2 [76], câncer [77], gota e aterosclerose [78].
Uma observação recente do grupo Rong Hu sobre a associação de Nrf2 com regulação negativa do inflamassoma revelou que, Nrf2 induz a expressão de NQO1 que leva à inibição da ativação do inflamassoma de NLRP3, clivagem da caspase-1 e IL-1? geração em macrófagos. Além disso, um ativador Nrf2 bem conhecido, a terc-butilhidroquinona (tBHQ) regulou negativamente a transcrição de NLRP3 ativando o ARE de maneira dependente de Nrf2 [79]. Além da observação acima, o mesmo grupo também foi revelado que o fumarato de dimetila (DMF) evita a colite induzida por DSS por meio da ativação da via de sinalização de Nrf2 que está envolvida na translocação nuclear de Nrf2 e na inibição da montagem do inflamassoma de NLRP3 [80].
Uma série de experimentos usando compostos naturais e sintéticos também revelou o efeito inibitório do Nrf2 na ativação do inflamassoma de NLRP3. Por exemplo, o tratamento de epigalocatequina-3-galato (EGCG) em camundongos com nefrite lúpica demonstrou diminuir a ativação do inflamassoma NLRP3 renal que é mediada pela via de sinalização Nrf2 [81]. Da mesma forma, citral (3,7-dimetil-2,6-octadienal), um composto ativo principal em um medicamento fitoterápico chinês Litsea cubeba, inibe a ativação do inflamassoma NLRP3 por meio da via de sinalização antioxidante Nrf2 em modelo de camundongo Nefrite Lúpica Grave e Acelerada (ASLN) [82]. Da mesma forma, a biochanina protegeu contra lesão hepática induzida por LPS / GalN ao ativar a via Nrf2 e inibir a ativação do inflamassoma de NLRP3 em camundongos BALB / c machos [83]. Além disso, a mangiferina também mostrou regular positivamente a expressão de Nrf2 e HO-1 de uma maneira dependente da dose e inibiu NLRP3 hepático induzido por LPS / D-GalN, ASC, caspase-1, IL-1? e TNF-? expressão [84].
Apesar da regulação negativa do NLRP3 pelo Nrf2, ele também ativa a função do inflamassoma NLRP3 e AIM2. Haitao Wen e colegas descobriram que, Nrf2? /? macrófagos de camundongo mostraram a ativação defeituosa do inflamassoma NLRP3 e AIM2, mas não do inflamassoma NLRC4 [85]. Curiosamente, esta observação descreve as funções desconhecidas do Nrf2 no contexto das doenças associadas à inflamação; portanto, é muito importante estudar mais para revelar o mecanismo pelo qual o Nrf2 ativa a função do inflamassoma antes de considerá-lo um alvo terapêutico.
Supressão da Transcrição de Citocinas Pró-Inflamatórias por Nrf2
Uma investigação muito recente baseada em resultados de imunoprecipitação da cromatina (ChIP) -seq e ChIP-qPCR em macrófagos de camundongo revelou que Nrf2 se liga às regiões promotoras de citocinas pró-inflamatórias, como IL-6 e IL-1? e inibe o recrutamento de RNA Pol II. Como resultado, o RNA Pol II é incapaz de processar a ativação transcricional de IL-6 e IL-1? isso, em última análise, leva à inibição da expressão gênica. Pela primeira vez, o grupo de Masayuki Yamamoto revelou o novo mecanismo pelo qual Nrf2 não apenas transativa seus genes downstream através de AREs, mas também suprime a ativação transcricional de genes específicos com ou sem ARE através da inibição do recrutamento de RNA Pol II [50].
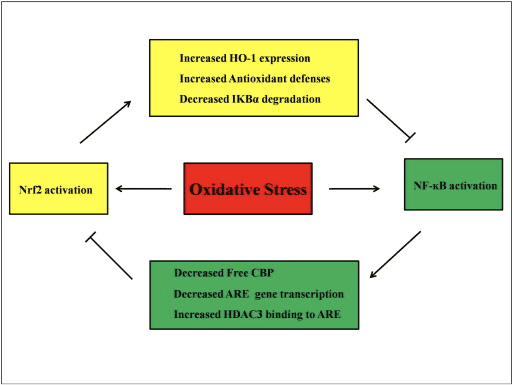
Conversa cruzada entre as vias Nrf2 e NF-? B
O NF-? B é um complexo proteico responsável pela transcrição do DNA encontrado em quase todos os tipos de células animais e envolvido em vários processos, como inflamação, apoptose, resposta imune, crescimento e desenvolvimento celular. p65, uma proteína Rel da família NF-? B, tem um domínio de transativação enquanto que p50 não possui e requer heterodimerização com a proteína Rel para ativar a transcrição. Durante o estresse oxidativo, a I? B quinase (IKK) é ativada e causa a fosforilação de I? B, resultando na liberação e translocação nuclear de NF-? B. O NF-? B causa a transcrição de mediadores pró-inflamatórios como IL-6, TNF- ?, iNOS, IL-1 e adesão intracelular COX-2.
A regulação anormal de NF-? B foi conectada à artrite reumatóide, asma, doença inflamatória intestinal e gastrite induzida por infecção por Helicobacter pylori [86]. Atualmente, considera-se que a atividade de NF-kB influencia a via de sinalização Keapl / Nrf2 / ARE principalmente em três aspectos: primeiro, Keap1 degrada IKK? através da ubiquitinação, inibindo assim a atividade do NF-? B [87]. Em segundo lugar, o processo inflamatório induz mediadores inflamatórios como COX2 derivado da ciclopentenona prostaglandina 15d-PGJ2, um eletrófilo forte que reage com Keap1 e ativa Nrf2, iniciando assim a transcrição do gene com inibição simultânea da atividade do NF-kB [58], [88] ( Fig. 3 A, B). Terceiro, o NF-? B pode se combinar com o coativador transcricional Nrf2 competitivo CBP [89], [90] (Fig. 3 C, D).
Figura 3 Diafonia entre as vias Nrf2 e NF-? B. (A) Keap1 direciona o IKK para a ubiquitinação mediada por CUL3 e degradação do proteassoma que leva à inibição da fosforilação de NF-? B e este mecanismo também funciona como ligação competitiva de Nrf2 e IKK com Keap1. (B) O estresse oxidativo ativa o IKK que fosforila o NF-? B, levando à sua translocação para o núcleo e ativação de citocinas pró-inflamatórias, como a COX-2. O produto terminal da COX-2 conhecido como 15d-PGJ2 atua como um indutor de Nrf2 que leva à supressão do estresse oxidativo. (C) Nrf2 se liga ao seu cofator de transcrição CBP junto com o pequeno Maf e outra maquinaria de transcrição para iniciar a expressão do gene dirigido por ARE. (D) Quando o NF-? B se liga ao CBP de maneira competitiva, ele inibe a ligação do CBP ao Nrf2, o que leva à inibição da transativação do Nrf2.
Assume-se que as vias de sinalização Nrf2 e NF-? B interagem para controlar a transcrição ou função de proteínas alvo a jusante. Na justificativa desta suposição, muitos exemplos mostram que a ativação e inibição direta ou indireta ocorrem entre os membros das vias Nrf2 e NF-? B (Fig. Em resposta ao LPS, o knockdown de Nrf2 aumenta significativamente a atividade transcricional de NF-? B e a transcrição do gene dependente de NF-? B, mostrando que Nrf2 impede a atividade de NF-? B [60], [91]. Além disso, a expressão aumentada de HO-2 dependente de Nrf1 a jusante inibe a atividade de NF-? B. Quando as células do câncer de próstata são brevemente expostas ao? -Tocoferil succinato, um derivado da vitamina E, a expressão de HO-1 é regulada positivamente. Os produtos finais de HO-1 inibem a translocação nuclear de NF-? B [92]. Estes estudos in vivo sugerem que o Nrf2 regula negativamente a via de sinalização do NF-kB. O LPS estimula a atividade de ligação ao DNA do NF-? B e o nível da subunidade p65 do NF-? B é significativamente maior em extratos nucleares dos pulmões de Nrf2? /? do que de camundongos WT, sugerindo um papel negativo de Nrf2 na ativação de NF-? B. Além disso, Nrf2? /? fibroblastos de embrião de camundongo tratados com LPS e TNF-? mostram uma ativação mais proeminente de NF-? B causada pela ativação de IKK e I? B-? degradação [60]. E a depuração do vírus sincicial respiratório é significativamente diminuída enquanto a atividade de ligação ao DNA do NF-? B é aumentada em Nrf2? /? ratos em comparação com ratos WT [93]. Nefrite lúpica induzida por pristane em Nrf2? /? camundongos co-tratados com sulforafano têm dano renal grave e alterações patológicas, bem como expressão elevada de iNOS e ativação de NF-? B em comparação com WT, sugerindo que Nrf2 melhora a nefrite lúpica ao inibir a via de sinalização de NF-? B e eliminar ROS [94 ] A atividade do NF-? B também ocorre quando as células são tratadas com um indutor Nrf2 juntamente com LPS e TNF- ?. Por exemplo, um derivado de chalcona sintético inibe a ativação de NF-? B induzida por TNF -? - tanto direta quanto indiretamente e parcialmente por meio da indução da expressão de HO-1 em células epiteliais HT-29 do intestino humano [62]. A supressão da translocação de NF-? B e da atividade de ligação ao DNA, bem como a supressão da expressão de iNOS em hepatócitos, são encontradas quando ratos F344 são tratados com 3H-1,2-ditiol-3-tiona (D3T) [95]. Após co-tratamento com sulforafano e LPS, a expressão induzida por LPS de iNOS, COX-2 e TNF-? em macrófagos crus 264.7 é regulado para baixo, sugerindo que o sulforafano tem atividade antiinflamatória por meio da inibição da ligação ao DNA de NF-? B [96]. Embora vários estudos experimentais tenham sido feitos para explicar a ligação entre as vias Nrf2 e NF-? B, resultados conflitantes permanecem. Regulamentos positivos e negativos foram relatados entre Nrf2 e NF-kB [97]. Normalmente, os eletrófilos quimiopreventivos 3H-1,2-ditiol-3-tiona, sulforafano e Triterpenóide CDDO-Me ativam o Nrf2 inibindo o NF-kB e seus genes regulados negativamente [98], [99], [100]. Em contraste, vários agentes ou condições como ROS, LPS, tensão de cisalhamento de fluxo, LDL oxidado e fumaça de cigarro mostraram aumentar a atividade de Nrf2 e NF-kB [97]. Além disso, estudos in vivo revelaram que a atividade de NF-kB está diminuída em fígados isolados de Nrf2? /? murganhos e a actividade de liga�o a NF-? B �mais baixa em Nrf2? /? do que em ratos Nrf2 + / + [101]. No entanto, as células endoteliais aórticas humanas tratadas com o vetor adenoviral Nrf2 inibem os genes a jusante do NF-? B sem afetar a atividade do NF-? B [8].
Figura 4 Loop regulatório de Nrf2 e NF-? B. A via de Nrf2 inibe a ativação de NF-? B, evitando a degradação de I? B-? e aumentando a expressão de HO-1 e as defesas antioxidantes que neutralizam ROS e produtos químicos desintoxicantes. Como resultado, a ativação de NF-? B associada a ROS é suprimida. Da mesma forma, a transcrição mediada por NF-? B reduz a ativação de Nrf2 reduzindo SOMOSTranscrição de gene e proteína de ligação CREB livre competindo com Nrf2 por CBP. Além disso, o NF-? B aumenta o recrutamento de histona desacetilase (HDAC3) para a região ARE e, portanto, a ativação transcricional de Nrf2 é evitada.
A ativação da via de sinalização Nrf2 desempenha um papel importante na expressão de enzimas e genes envolvidos na desintoxicação de oxidantes reativos, aumentando a capacidade antioxidante das células no corpo humano. Enquanto muitos estudos de pesquisa estão disponíveis hoje, os mecanismos regulatórios na ativação do Nrf2 não são totalmente compreendidos. Um possível papel da via de sinalização Nrf2 no tratamento da inflamação também foi encontrado. Dr. Alex Jimenez DC, Insight CCST
Papel do Nrf2 nas Doenças Inflamatórias
Estudos in vivo mostraram que o Nrf2 desempenha um papel importante em doenças inflamatórias que afetam diferentes sistemas; Estes incluem gastrite, colite, artrite, pneumonia, danos no fígado, doenças cardiovasculares, doenças neurodegenerativas e lesões cerebrais. Nestes estudos, Nrf2? /? os animais mostraram sintomas mais graves de inflamação e danos nos tecidos do que os animais WT. Portanto, acredita-se que a via de sinalização Nrf2 tenha um efeito protetor nas doenças inflamatórias. A instalação intra-traqueal de elastase pancreática porcina induz doença pulmonar obstrutiva crônica, particularmente enfisema. Os camundongos deficientes em Nrf2 são altamente suscetíveis ao enfisema, e a diminuição da expressão de HO-1, PrxI e do gene antiprotease SLPI ocorre em macrófagos alveolares. O Nrf2 é considerado um regulador chave no sistema de defesa mediado por macrófagos contra a lesão pulmonar [102]. Camundongos deficientes em Nrf2 com enfisema induzido pela exposição ao fumo do tabaco nos meses 6 mostram aumento da inflamação broncoalveolar, expressão aumentada de marcadores de estresse oxidativo nos alvéolos e aumento da apoptose de células septais alveolares, sugerindo que o Nrf2 atua contra o enfisema induzido pelo tabaco genes [102], [103]. Com disrupção Nrf2, a inflamação das vias aéreas mediada por alérgenos e asma usando complexo ovalbumina mostram aumento da inflamação das vias aéreas, hiper-reatividade das vias aéreas, hiperplasia de células caliciformes e altos níveis de Th2 no lavado broncoalveolar e esplenócitos, enquanto a via de sinalização mediada por Nrf2 limita a eosinofilia nas vias aéreas hipersecreção de muco e hiper-reatividade das vias aéreas, além de induzir muitos genes antioxidantes que impedem o desenvolvimento de asma [104]. A injeção de carragenina na cavidade pleural induz a pleurisia e o acúmulo de 15d-PGJ2 nas células inflamatórias Nrf2 está confinado aos macrófagos peritoneais de camundongos. Durante a fase inicial da inflamação, 15d-PGJ2 ativa Nrf2 e regula o processo inflamatório através da indução de HO-1 e PrxI. Um estudo também sugeriu que COX-2 tem um efeito anti-inflamatório na fase inicial pela produção de 15d-PGJ2 [105]. A administração oral de 1% dextran sulfato de sódio na semana 1 induz a colite associada a alterações histológicas que incluem encurtamento das criptas e infiltração de células inflamatórias no tecido do cólon. Para proteger a integridade intestinal da colite, o Nrf2 pode desempenhar um papel importante regulando as citocinas pró-inflamatórias e induzindo enzimas desintoxicantes de fase II [51]. Em um modelo de camundongo nocaute para Nrf2 de sepse pulmonar induzida por LPS, a atividade de NF-? B regula a influência de citocinas inflamatórias, como COX-2, IL-113, IL-6 e TNF? que são essenciais para iniciar e promover a inflamação [60]. Nrf2 reduz o dano inflamatório, regulando esses fatores inflamatórios. Nesses modelos de inflamação aguda, o aumento da regulação de enzimas antioxidantes, citocinas pró-inflamatórias e mediadores pela via de sinalização Nrf2 reduz a lesão inflamatória em animais WT. Curiosamente, isso também foi relatado em camundongos Nrf2-knockout em que os sintomas são acentuadamente exacerbados em comparação com camundongos WT.
Pesquisa sobre drogas antiinflamatórias dependentes de Nrf2
Em resumo, discutimos expericias mostrando que a via do sinal Nrf2 desempenha um papel regulador em muitas eas de inflamao, pelo que os agentes anti-inflamatios dependentes de Nrf2 s importantes para o tratamento de doens inflamatias.
As plantas têm sido fontes extraordinariamente ricas de compostos que ativam o fator de transcrição Nrf2, levando à regulação positiva de genes citoprotetores. Recentemente, vários estudos foram conduzidos para investigar os efeitos de diferentes agentes anti-inflamatórios, principalmente de origem vegetal. Por exemplo, a curcumina é o ingrediente ativo da cúrcuma e também é encontrada em pequenas quantidades no gengibre; isotiocianatos, especificamente fenilisotiocianatos são de brócolis, aipo e outros vegetais; e antocianinas são de bagas e uvas [124]. Estudos demonstraram que todos esses agentes não são apenas bons antioxidantes, mas também têm potentes efeitos anti-inflamatórios através da indução de Nrf2 [125], [126]. Portanto, o desenvolvimento de novos ativadores antiinflamatórios Nrf2 a partir de extrato vegetal tem atraído muito interesse em pesquisas médicas.
Nos últimos anos, muitos experimentos com animais foram realizados para confirmar as ações desses compostos. Artesunato é usado principalmente para malária grave, malária cerebral e doenças reumáticas autoimunes; também é eficaz em lesões pulmonares sépticas. O artesunato ativa a expressão de Nrf2 e HO-1, e o último reduz o influxo de citocinas pró-inflamatórias e leucócitos no tecido para prevenir a inflamação [127]. Acredita-se que a isovitexina, extraída da casca do arroz Oryza sativa, tenha propriedades antiinflamatórias e antioxidantes; ele desempenha um papel protetor contra a lesão pulmonar aguda induzida por LPS, ativando a via Nrf2 / HO-1 e inibindo MAPK e NF-? B [128]. Fimasartan, um bloqueador do receptor da angiotensina II recentemente popular que atua no sistema renina-angiotensina, reduz a pressão arterial; o uso de fimasartan para tratar camundongos com obstrução ureteral unilateral induzida cirurgicamente reduz o estresse oxidativo, inflamação e fibrose por meio da regulação positiva de Nrf2 e da via antioxidante e da inibição de RAS e MAPKs [129]. A sappanona é amplamente distribuída no sudeste da Ásia, onde é usada como medicamento anti-influenza, anti-alérgico e neuroprotetor; ativa o Nrf2 e inibe o NF-? B e, portanto, pode ser benéfico no tratamento de doenças relacionadas ao Nrf2- e / ou NF-? B [130]. Bixin extraído das sementes de Bixin orellana é usado para doenças infecciosas e inflamatórias no México e na América do Sul; diminui os mediadores inflamatórios, o vazamento capilar alveolar e o dano oxidativo de uma maneira dependente de Nrf2 para aliviar a lesão pulmonar induzida pela ventilação e restaurar a morfologia pulmonar normal [131]. Outros compostos de plantas, como galato de epigalocatequina, sulforafano, resveratrol, licopeno e extrato de chá verde têm efeitos terapêuticos em doenças inflamatórias por meio da via de sinalização Nrf2 [132], [133], [134]. Recentemente, outro fitoquímico, eriodictiol, que está presente em frutas cítricas, foi relatado por ter efeitos antiinflamatórios e antioxidantes na lesão renal induzida por cisplatina e lesão pulmonar aguda induzida por sepse ao regular Nrf2, inibir NF-? B e inibir a expressão de citocinas em macrófagos [135], [136]. No entanto, vários fitoquímicos mostram grande promessa para a prevenção e tratamento de várias doenças humanas, e alguns já entraram na fase de ensaios clínicos (Tabela 2).
Estes compostos vegetais ativam a via de sinalização Nrf2 principalmente na forma de materiais eletrofílicos que modificam os resíduos de cisteína do Keap1, levando à ligação nuclear livre de Nrf2 com o ARE, resultando na ativação da transcrição do gene correspondente.
Sulforafano e seus efeitos sobre câncer, mortalidade, envelhecimento, cérebro e comportamento, doenças cardíacas e muito mais
Os isotiocianatos são alguns dos compostos vegetais mais importantes que você pode obter em sua dieta. Neste vídeo eu faço o caso mais abrangente para eles que já foi feito. Curto período de atenção? Pule para o seu tópico favorito clicando em um dos pontos de tempo abaixo. Cronograma completo abaixo.
Seções principais:
00: 01: 14 - Câncer e mortalidade
00: 19: 04 - envelhecimento
00: 26: 30 - Cérebro e comportamento
00: 38: 06 - recapitulação final
00: 40: 27 - dose
Cronograma completo:
00: 00: 34 - Introdução do sulforafano, um dos principais focos do vídeo.
00: 01: 14 - Consumo de vegetais crucíferos e reduções na mortalidade por todas as causas.
00: 02: 12 - risco de câncer de próstata.
00: 02: 23 - risco de câncer de bexiga.
00: 02: 34 - Câncer de pulmão em risco de fumantes.
00: 02: 48 - risco de câncer de mama.
00: 03: 13 - Hipotético: e se você já tem câncer? (intervencionista)
00: 03: 35 - Mecanismo plausível que direciona os dados associativos de câncer e mortalidade.
00: 04: 38 - Sulforafano e câncer.
00: 05: 32 - Evidência animal mostrando forte efeito do extrato de brócolis no desenvolvimento do tumor de bexiga em ratos.
00: 06: 06 - Efeito da suplementação direta de sulforafano em pacientes com câncer de próstata.
00: 07: 09 - Bioacumulação de metabólitos de isotiocianato no tecido mamário atual.
00: 08: 32 - Inibição de células estaminais de cancro da mama.
00: 08: 53 - Lição de História: os brassicas foram estabelecidos como tendo propriedades de saúde mesmo na Roma antiga.
00: 09: 16 - A capacidade do Sulforaphane de aumentar a excreção de carcinógeno (benzeno, acroleína).
00: 09: 51 - NRF2 como um interruptor genético através de elementos de resposta antioxidante.
00: 10: 10 - Como a ativação de NRF2 aumenta a excreção de carcinógenos via conjugados de glutationa-S.
00: 10: 34 - As couves-de-bruxelas aumentam a glutationa-S-transferase e reduzem os danos no DNA.
00: 11: 20 - Bebida de brócolis aumenta a excreção de benzeno em 61%.
00: 13: 31 - O homogenato de brócolis aumenta as enzimas antioxidantes nas vias aéreas superiores.
00: 15: 45 - Consumo de vegetais crucíferos e mortalidade por doenças cardíacas.
00: 16: 55 - Brócolis em pó melhora os lipídios no sangue e o risco geral de doenças cardíacas em diabéticos tipo 2.
00: 19: 04 - Início da seção de envelhecimento.
00: 19: 21 - dieta enriquecida com sulforafano aumenta a vida útil de besouros de 15 a 30% (em certas condições).
00: 20: 34 - Importância da baixa inflamação para a longevidade.
00: 22: 05 - Os vegetais crucíferos e o pó de brócolis parecem reduzir uma grande variedade de marcadores inflamatórios em humanos.
00: 23: 40 - Recapitulação de vídeo intermediário: câncer, seções de envelhecimento
00: 24: 14 - Estudos com ratos sugerem que o sulforafano pode melhorar a função imunológica adaptativa na velhice.
00: 25: 18 - Sulforaphane melhorou o crescimento do cabelo em um modelo de rato de calvície. Imagem no 00: 26: 10.
00: 26: 30 - Início da seção do cérebro e comportamento.
00: 27: 18 - Efeito do extrato de brócolis no autismo.
00: 27: 48 - Efeito da glucorafanina na esquizofrenia.
00: 28: 17 - Início da discussão sobre depressão (mecanismo plausível e estudos).
00: 31: Estudo 21 - Mouse usando 10 diferentes modelos de depressão induzida por estresse mostram sulforafano igualmente eficaz como fluoxetina (prozac).
00: 32: 00 - Estudo mostra a ingestão direta de glucorafanina em camundongos é igualmente eficaz na prevenção da depressão do modelo de estresse de derrota social.
00: 33: 01 - Início da seção de neurodegeneração.
00: 33: 30 - Sulforafano e doença de Alzheimer.
00: 33: 44 - Sulforaphane e doença de Parkinson.
00: 33: 51 - Sulforaphane e doença de Hungtington.
00: 34: 13 - Sulforafano aumenta as proteínas de choque térmico.
00: 34: 43 - Início da seção de traumatismo cranioencefálico.
00: 35: 01 - Sulforafano injetado imediatamente após o TBI melhora a memória (estudo do mouse).
00: 35: 55 - Sulforafano e plasticidade neuronal.
00: 36: 32 - Sulforaphane melhora o aprendizado em modelos de diabetes tipo II em camundongos.
00: 37: 19 - Sulforafano e distrofia muscular de duchenne.
00: 37: 44 - Inibição da miostatina em células satélites musculares (in vitro).
00: 38: 06 - Recapitulação de vídeo tardio: mortalidade e câncer, danos no DNA, estresse oxidativo e inflamação, excreção de benzeno, doença cardiovascular, diabetes tipo II, efeitos no cérebro (depressão, autismo, esquizofrenia, neurodegeneração), via NRF2.
00: 40: 27 - Pensamentos em descobrir uma dose de brotos de brócolis ou sulforafano.
00: 41: 01 - Anedotas sobre brotar em casa.
00: 43: 14 - Nas temperaturas de cozimento e atividade de sulforafano.
00: 43: 45 - Conversão da bactéria intestinal do sulforafano da glucorafanina.
00: 44: 24 - Os suplementos funcionam melhor quando combinados com a mirosinase ativa de vegetais.
00: 44: 56 - Técnicas de cozinha e vegetais crucíferos.
00: 46: 06 - Isotiocianatos como sendo goitrogénios.
Conclusões
Atualmente, muitas pesquisas têm se concentrado no papel da via de sinalização Nrf2 / Keap1 / ARE na inflamação. Entre as enzimas reguladas positivamente por Nrf2, HO-1 é uma das enzimas de resposta ao estresse representativas. HO-1 tem propriedades antiinflamatórias e antioxidantes proeminentes. Em geral, a via de sinalização Nrf2 também regula negativamente citocinas, fatores de liberação de quimiocinas, MMPs e outros mediadores inflamatórios COX-2 e produção de iNOS, que afetam direta ou indiretamente as vias NF-kB e MAPK relevantes e outras redes que controlam a inflamação. É sugerido que as vias de sinalização de Nrf2 e NF-? B interagem para regular a transcrição ou função de proteínas alvo a jusante. A supressão ou inativação da atividade transcricional mediada pelo NF-? B através do Nrf2 ocorre muito provavelmente na fase inicial da inflamação, uma vez que o NF-? B regula a síntese de novo de uma série de mediadores pró-inflamatórios. No entanto, ainda existem algumas limitações na pesquisa, como se há conexões entre Nrf2 e outras vias de sinalização, como JAK / STAT, a importância dos ativadores Nrf2 atuais derivados de fontes naturais de plantas na inflamação e como melhorar a atividade biológica e aumentar o direcionamento desses compostos. Estes requerem validação experimental adicional.
Além disso, a via de sinalização Nrf2 pode regular> 600 genes [163], dos quais> 200 codificam proteínas citoprotetoras [164] que também estão associadas a inflamação, câncer, doenças neurodegenerativas e outras doenças importantes [165]. Evidências crescentes sugerindo que a via de sinalização do Nrf2 está desregulada em muitos cânceres, resultando em uma bateria de genes dependentes de Nrf2 de expressão aberrante. Além disso, a inflamação desempenha um papel importante nas doenças relacionadas ao estresse oxidativo, especialmente no câncer. A aplicação de vários ativadores Nrf2 para neutralizar a inflamação pode resultar na expressão aberrante de genes Nrf2 a jusante que induz a oncogênese e resistência à quimioterapia e / ou radioterapia. Portanto, ativadores altamente específicos de Nrf2 podem ser desenvolvidos para minimizar seus efeitos pleiotrópicos. Vários ativadores de Nrf2 mostraram uma melhora significativa das funções antiinflamatórias em doenças relacionadas ao estresse oxidativo. O melhor exemplo de ativador Nrf2 aprovado pelo FDA e amplamente utilizado para o tratamento de doenças inflamatórias, como esclerose múltipla (EM), é o fumarato de dimetila. Tecfidera (nome registrado de fumarato de dimetila pela Biogen) usado efetivamente para tratar formas recorrentes de esclerose múltipla em um grande número de pacientes [152]. No entanto, a eficácia do uso de ativadores Nrf2 para tratar doenças inflamatórias requer validação adicional para evitar os efeitos deletérios do Nrf2. Portanto, o desenvolvimento de terapias para a atividade antiinflamatória mediada por Nrf2 pode ter impacto clínico significativo. Os estudos em andamento da via de sinalização do Nrf2 em todo o mundo são dedicados ao desenvolvimento de agentes terapêuticos altamente direcionados para controlar os sintomas da inflamação e prevenir e tratar o câncer, bem como doenças neurodegenerativas e outras doenças importantes.
Em conclusão, o Nrf2 detecta os níveis de estresse oxidativo no corpo humano e, finalmente, ajuda a promover a regulação de enzimas e genes antioxidantes e desintoxicantes. Porque a inflamação crônica causada pelo aumento dos níveis de estresse oxidativo tem sido associada a doenças neurodegenerativas, Nrf2 pode desempenhar um papel essencial no tratamento de questões de saúde como a doença de Alzheimer, entre outras. O escopo de nossas informações é limitado a questões de quiropraxia e saúde da coluna vertebral. Para discutir o assunto, sinta-se à vontade para perguntar ao Dr. Jimenez ou entre em contato conosco em 915-850-0900 .
Discussão Adicional do Tópico: Aliviar a Dor no Joelho sem Cirurgia
A dor no joelho é um sintoma bem conhecido que pode ocorrer devido a uma variedade de lesões e / ou condições no joelho, incluindo lesões esportivas. O joelho é uma das articulações mais complexas do corpo humano, pois é formado pela intersecção de quatro ossos, quatro ligamentos, vários tendões, dois meniscos e cartilagem. De acordo com a Academia Americana de Médicos de Família, as causas mais comuns de dor no joelho incluem subluxação patelar, tendinite patelar ou joelho de saltador e doença de Osgood-Schlatter. Embora a dor no joelho seja mais provável de ocorrer em pessoas com mais de 60 anos, a dor no joelho também pode ocorrer em crianças e adolescentes. Dor no joelho pode ser tratada em casa seguindo os métodos RICE; no entanto, lesões graves no joelho podem exigir atenção médica imediata, incluindo cuidados quiropráticos.
Doenças neurodegenerativas, como a doença de Alzheimer e a doença de Parkinson, afetam milhões de pessoas em todo o mundo. Uma variedade de opções de tratamento está disponível para tratar os sintomas de várias doenças neurodegenerativas, embora os resultados sejam frequentemente limitados. Estudos de pesquisa descobriram que o estresse oxidativo causado por fatores internos e externos pode ser uma causa para o desenvolvimento de doenças neurodegenerativas. o fator de transcrição, Nrf2, foi determinado para funcionar como um importante mecanismo de defesa contra o estresse oxidativo. O objetivo do artigo abaixo é mostrar os efeitos de Nrf2 em doenças neurodegenerativas.
Modulação da Proteostase pelo Fator de Transcrição NRF2
As doenças neurodegenerativas estão ligadas ao acúmulo de agregados proteicos específicos, sugerindo uma íntima conexão entre o cérebro lesado e a perda de proteostase. Proteostasis refere-se a todos os processos pelos quais as células controlam a abundância e dobramento do proteoma graças a uma ampla rede que integra a regulação de vias de sinalização, expressão gênica e sistemas de degradação de proteínas. Esta revisão tenta resumir os achados mais relevantes sobre a modulação transcricional da proteostase exercida pelo fator de transcrição NRF2 (fator nuclear (2 derivado de eritrócitos), como 2). O NRF2 foi considerado classicamente como o principal regulador da resposta das células antioxidantes, embora esteja emergindo atualmente como um componente-chave da maquinaria de transdução para manter a proteostase. Como discutiremos, o NRF2 poderia ser visualizado como um hub que compila os sinais de emergência derivados do acúmulo de proteínas desdobradas, a fim de construir uma resposta de transcrição coordenada e perdurável. Isso é conseguido pelas funções do NRF2 relacionadas ao controle de genes envolvidos na manutenção da fisiologia do retículo endoplasmático, do proteassomo e da autofagia.
O fator nuclear (derivado do eritroide 2) -like 2 (NRF2) é uma proteína zipper de leucina básica considerada hoje em dia como um regulador mestre da homeostase celular. Ele controla a expressão basal e induzível por estresse de mais de 250 genes que compartilham em comum um intensificador de ação cis denominado elemento de resposta antioxidante (ERA) [1], [2], [3], [4], [5]. Esses genes participam das reações de desintoxicação de fase I, II e III, metabolismo da glutationa e da peroxirredoxina / tiorredoxina, produção de NADPH pela via da pentose fosfato e enzima málica, oxidação de ácidos graxos, metabolismo do ferro e proteostase [6]. Dadas essas amplas funções citoprotetoras, é possível que um único acerto farmacológico no NRF2 possa mitigar o efeito dos principais culpados de doenças crônicas, incluindo estresse oxidativo, inflamatório e proteotóxico. O papel do NRF2 na modulação da defesa antioxidante e resolução da inflamação foi abordado em vários estudos (revisado em [7]). Aqui, vamos nos concentrar em seu papel na proteostase, ou seja, no controle homeostático da síntese, dobramento, tráfego e degradação de proteínas. Exemplos serão fornecidos no contexto de doenças neurodegenerativas.
Perda de Proteostase Influencia Atividade NRF2 em Doenças Neurodegenerativas
Uma característica geral das doenças neurodegenerativas é a ocorrência de agregação aberrante de algumas proteínas. Assim, agregados de proteínas mal dobrados de? -Sinucleína (? -SYN) são encontrados na doença de Parkinson (PD), placas? -Amilóides (A?) E emaranhados neurofibrilares TAU hiperfosforilados na doença de Alzheimer (AD), huntingtina (Htt) em Doença de Huntington (HD), superóxido dismutase 1 (SOD1) e proteína de ligação de DNA TAR 43 (TDP-43) na esclerose lateral amiotrófica (ALS), proteína príon (PrP) em encefalopatias espongiformes, etc. Os agregados de proteína podem ter um impacto em vários vias celulares, que por sua vez podem afetar os níveis e a atividade de NRF2.
Diferentes camadas de regulação controlam firmemente a atividade NRF2
Sob condições fisiológicas, as células exibem baixos níveis de proteína NRF2 devido ao seu rápido turnover. Em resposta a diferentes estímulos, a proteína NRF2 é acumulada, entra no núcleo e aumenta a transcrição de genes contendo ARE. Portanto, o gerenciamento dos níveis de proteína NRF2 é um ponto chave que deve integrar sinais de entrada positivos e negativos. Como discutiremos mais adiante, o NRF2 é ativado por diversos mecanismos de sobreposição para orquestrar uma resposta rápida e eficiente, mas, por outro lado, o NRF2 pode ser inibido, provavelmente em uma segunda fase, a fim de desligar sua resposta.
Do ponto de vista clássico, a ativação do NRF2 tem sido considerada uma consequência da resposta celular a compostos oxidantes ou eletrofílicos. A este respeito, a proteína 3 associada a ECH semelhante a Kelch do adaptador de ubiquitina E1 ligase (KEAP1) desempenha um papel crucial. Os detalhes moleculares serão tratados mais detalhadamente na Seção 4.1. Em resumo, o KEAP1 atua como um sensor redox devido aos resíduos de cisteína críticos que levam à ubiquitinação do NRF2 e à degradação do proteassoma. Além dessa modulação clássica, o NRF2 é profundamente regulado por eventos de sinalização. Na verdade, foi demonstrado que diferentes quinases fosforilam e regulam o NRF2. Por exemplo, NRF2 pode ser fosforilado por proteínas quinases ativadas por mitogênio (MAPKs), embora sua contribuição para a atividade de NRF2 permaneça obscura [8], [9], [10], [11]. PKA quinase, bem como algumas isoenzimas PKC [12], CK2 [13] ou Fyn [14] fosforilam NRF2 modificando sua estabilidade. Trabalhos anteriores de nosso grupo relataram que a glicogênio sintase quinse-3? (GSK-3?) Inibe NRF2 por exclusão nuclear e degradação proteassomal [15], [25], [26], [27], [28], [29], [30]. Os detalhes moleculares serão discutidos na Seção 4.1. Além disso, a NRF2 está submetida a outros tipos de regulamentação. Por exemplo, a acetilação de NRF2 por CBP / p300 aumenta sua atividade [17], enquanto é inibida por miR153, miR27a, miR142-5p e miR144 [16], ou por metilação de ilhas de citosina-guanina (CG) dentro do promotor NRF2 [18].
Impacto de Agregados Proteicos nos Mecanismos Regulamentares NRF2
Nesta seção, vamos nos concentrar em como o acúmulo de proteínas com dobramento incorreto pode afetar a atividade de NRF2, fornecendo alguns dos caminhos mencionados acima como exemplos ilustrativos. Em primeiro lugar, precisamos considerar que o acúmulo de proteínas está intimamente ligado ao dano oxidativo. De fato, o acúmulo e a agregação incorreta de proteína induzidos induzem a produção anormal de espécies reativas de oxigênio (ROS) das mitocôndrias e de outras fontes [19]. Como mencionado acima, o ROS modificará as cisteínas sensíveis ao redox do KEAP1, levando à liberação, estabilização e localização nuclear do NRF2.
Em relação às proteinopatias, um exemplo de eventos de sinalização desregulados que podem afetar o NRF2 é fornecido pela hiperativação de GSK-3? em anúncio. GSK-3 ?, também conhecida como TAU quinase, participa da fosforilação dessa proteína associada aos microtúbulos, resultando em sua agregação, formação de emaranhados neurofibrilares e interrupção do transporte axonal (revisado em [20]). Por outro lado, GSK-3? reduz drasticamente os níveis e a atividade de NRF2 como mencionado acima. Embora não seja amplamente aceita, a cascata amilóide propõe que o A? oligômeros aumentam GSK-3? atividade junto com a hiperfosforilação TAU e morte de neurônios [21], [22]. Existem diferentes modelos para explicar como A? favorece GSK3-? atividade. Por exemplo, A? liga-se ao receptor de insulina e inibe as vias de sinalização de PI3K e AKT, que são cruciais para manter GSK-3? inativado por fosforilação em seu resíduo Ser9 N-terminal [23]. Por outro lado, extracelular A? interage com os receptores Frizzled, bloqueando a sinalização WNT [24] e novamente resultando na liberação de GSK-3 ?. Em resumo, A? o acúmulo leva à hiperativação anormal de GSK-3 ?, prejudicando assim uma resposta de NRF2 apropriada.
Como discutido na seção a seguir, as proteínas com dobras incorretas levam à ativação de PERK e MAPKs, que por sua vez regulam NRF2 [31], [8], [9], [10], [11]. Além disso, a atividade desregulada de CBP / p300 foi relatada em várias proteinopatias [32] e uma diminuição global na metilação do DNA em cérebros com DA também foi mostrada [33], fornecendo, portanto, fundamentos para explorar a relevância desses achados na regulação de NRF2.
Nós e outros observaram em necrópsias de pacientes com DP e DA um aumento nos níveis de proteína NRF2 e alguns de seus alvos, como heme oxigenase 1 (HMOX1), NADPH quinona oxidase 1 (NQO1), p62, etc., ambos por immunoblot e por imuno-histoquímica [34], [35], [36], [37], [38], [39]. A regulação positiva de NRF2 nessas doenças é interpretada como uma tentativa malsucedida do cérebro doente de recuperar os valores homeostáticos. No entanto, outro estudo indicou que NRF2 é predominantemente localizado no citoplasma dos neurônios do hipocampo AD, sugerindo redução da atividade transcricional de NRF2 no cérebro [40]. É concebível que a disparidade dessas observações esteja relacionada a mudanças nos fatores que controlam o NRF2 ao longo dos estágios progressivos da neurodegeneração.
Três sistemas principais contribuem para a proteostase, ou seja, a resposta protéica desdobrada (UPR), o sistema de proteassoma ubiquitina (UPS) e autofagia. Em seguida, apresentamos evidências para visualizar NRF2 como um hub conectando sinais de emergência ativados por agregados de proteína com o maquinário derivativo de proteína.
NRF2 participa da resposta protéica desdobrada (UPR)
Ativação NRF2 em resposta ao UPR
O dobramento de proteína oxidativa no ER é conduzido por uma série de vias distintas, a mais conservada das quais envolve a proteína dissulfeto-isomerase (PDI) e a sulfidril oxidase endoplasmática oxidoredutina 1 (ERO1? E ERO1? Em mamíferos) como doador de dissulfeto. Resumidamente, o PDI catalisa a formação e quebra de ligações dissulfeto entre resíduos de cisteína dentro das proteínas, conforme elas se dobram, devido à redução e oxidação de seus próprios aminoácidos de cisteína. O PDI é reciclado pela ação da enzima doméstica ERO1, que reintroduz ligações dissulfeto em PDI [41]. O oxigênio molecular é o aceptor terminal de elétrons de ERO1, que gera quantidades estequiométricas de peróxido de hidrogênio para cada ligação dissulfeto produzida [42]. Peroxidases (PRX4) e glutationa peroxidases (GPX7 e GPX8) são enzimas chave para reduzir o peróxido de hidrogênio no RE. Quando este sistema redutor de óxido não funciona adequadamente, o acúmulo anormal de proteínas mal dobradas ocorre no ER e um conjunto de sinais denominado resposta da proteína não dobrada (UPR) é transmitido ao citoplasma e ao núcleo para restabelecer a homeostase do ER [43]. Três proteínas associadas à membrana foram identificadas para detectar estresse ER em eucariotos: fator de transcrição de ativação 6 (ATF6), ER pancreático eIF2? quinase (PERK, também proteína quinase tipo ER quinase ativada por RNA de fita dupla) e quinase 1 exigindo inositol (IRE1). O domínio luminal de cada sensor está ligado a uma chaperona de 78 kDa denominada proteína regulada por glicose (GRP78 / BIP). O BIP se dissocia com o estresse do ER para ligar as proteínas não dobradas, levando à ativação dos três sensores [44].
O NRF2 e o seu homólogo NRF1, também relacionados à resposta antioxidante, participam na transdução do UPR para o núcleo. No caso de NRF1, esta proteína está localizada na membrana do RE e sofre translocação nuclear após desglicosilação ou clivagem. Então, a ativação de UPR leva ao processamento de NRF1 e ao acúmulo nuclear do fragmento resultante no compartimento nuclear. No entanto, a capacidade de transativar genes contendo ARE deste fragmento NRF1 ainda está em discussão [45].
Glover-Cutter e colaboradores mostraram a ativação do ortólogo NRF2 de C. elegans, SKN-1, com diferentes estressores ER. O aumento da expressão de SKN-1 foi dependente de diferentes mediadores de UPR, incluindo os ortólogos de vermes IRE1 ou PERK [46]. Em células deficientes em PERK, a síntese proteica deficiente leva ao acúmulo de peróxidos endógenos e subsequente apoptose [47]. O efetor usado pela PERK para proteger o ER desses peróxidos pode ser NRF2, uma vez que foi reportado que a PERK fosforila NRF2 no Ser40, evitando assim sua degradação pelo KEAP1 [31]. A indução de ASK1 também é susceptível de desempenhar um papel nesta via através da acção da quinase mediada por TRAF2 de IRE1 [48]. Embora o papel das MAPKs na regulação de NRF2 ainda seja controverso, foi recentemente sugerido que a via IRE1-TRAF2-ASK1-JNK poderia ativar NRF2 [49] (Fig. 1). Curiosamente, em C. elegans e células humanas, novas evidências sugerem que a sulfenilação de cisteína da IRE1 quinase na sua alça de ativação inibe a UPR mediada por IRE1 e inicia uma resposta antioxidante p38 conduzida por NRF2. Os dados sugerem que IRE1 tem uma função antiga como sentinela citoplasmática que ativa p38 e NRF2 [50].
Figura 1 Regulamento de NRF2 pela UPR. O acúmulo de proteínas desdobradas ou mal dobradas dentro do retículo endoplasmático pode iniciar a resposta protéica desdobrada (RPU). Em primeiro lugar, o BIP chaperona é liberado do domínio intraluminal dos sensores ER ERRENUMX e PERK para ligar proteínas desdobradas / desdobradas. Isto permite a dimerização e a trans-auto-fosforilação dos seus domínios citosólicos. A ativação de PERK resulta em fosforilação direta de NRF1 em Ser2, levando à translocação NRF40 para o núcleo e ativação de genes alvo. A ativação de IRE2 induz o recrutamento de TRAF1 seguido pela fosforilação e ativação de ASK2 e JNK. Como foi relatado que o JNK fosforila e ativa o NRF1, é razoável pensar que a ativação do IRE2 levaria ao aumento da atividade do NRF1.
Muitos estudos sobre a indução da UPR foram conduzidos com o inibidor da glicosilação de proteínas tunicamicina. NRF2 parece ser essencial para a prevenção da morte celular apoptótica induzida por tunicamicina [31] e sua ativação sob essas condições é conduzida pela degradação autofágica de KEAP1 [51]. Consequentemente, o silenciamento mediado por shRNA da expressão de NRF2 em células? TC-6, uma linha de células de insulinoma? De murino, aumentou significativamente a citotoxicidade induzida por tunicamicina e levou a um aumento na expressão do marcador de estresse ER pró-apoptótico CHOP10. Por outro lado, a ativação do NRF2 por 1,2-ditiol-3-tiona (D3T) reduziu a citotoxicidade da tunicamicina e atenuou a expressão de CHOP10 e PERK [52]. Curiosamente, os neurônios olfatórios submetidos à aplicação sistêmica de tunicamicina aumentaram o NRF2 em paralelo com outros membros da UPR, como CHOP, BIP, XBP1 [53]. Estes resultados foram estendidos a estudos in vivo, uma vez que a infusão ventricular lateral de tunicamicina em ratos induziu a expressão de PERK e NRF2 no hipocampo acompanhada por déficits cognitivos significativos, aumento da fosforilação de TAU e depósitos de A? 42 [54].
NRF2 Up-Regula Genes Chave para a Manutenção da Fisiologia do ER
O lúmen ER precisa de um suprimento abundante de GSH do citosol para manter a química do dissulfeto. NRF2 modula enzimas cruciais do metabolismo de GSH no cérebro, como transporte de cistina / glutamato,? -Glutamato cisteína sintetase (? -GS), glutamato-cisteína ligase catalítica e subunidades moduladoras (GCLC e GCLM), glutationa redutase (GR) e glutationa peroxidase (GPX) (revisado em [55]). A relevância de NRF2 na manutenção de GSH no ER é suportada pela descoberta de que a ativação farmacológica ou genética de NRF2 resulta no aumento da síntese de GSH via GCLC / GCLM, enquanto a inibição da expressão dessas enzimas por NRF2-knockdown causou um acúmulo de danos proteínas dentro do ER levando à ativação UPR [56].
Em C. elegans, vários componentes dos genes alvo UPR regulados por SKN-1, incluindo Ire1, Xbp1 e Atf6. Embora NRF2 regule positivamente a expressão de vários genes de peroxidase (PRX) e glutationa peroxidase (GPX) em mamíferos (revisado em [57]), apenas GPX8 é uma enzima genuína localizada no ER, abrigando o sinal de recuperação KDEL [58]. A perda de GPX8 causa ativação de UPR, vazamento de peróxido de hidrogênio derivado de ERO1 <1> para o citosol e morte celular. Peróxido de hidrogênio derivado de ERO8? a atividade não pode se difundir do ER para o citosol devido à ação combinada de GPX4 e PRX59 [2]. A este respeito, uma análise do arranjo de expressão do gene da via de defesa antioxidante usando RNA de tecido de camundongo do tipo selvagem e NRF8-null, revelou que a expressão de GPX2 foi regulada negativamente na ausência de NRF60 [2]. Em consonância com isso, a análise do transcriptoma de amostras de pacientes que sofrem de neoplasias mieloproliferativas, policitemia ou mielofibrose, doenças também associadas com estresse oxidativo e inflamação crônica de baixo grau, mostram níveis de expressão mais baixos de NRF8 e GPX61 em comparação com indivíduos de controle [8]. Ainda não há estudos que envolvam especificamente o GPX8 na proteção do cérebro humano, mas uma análise do transcriptoma em camundongos indica um aumento compensatório do GPX62 na resposta à toxina parkinsoniana MPTP [XNUMX].
Impacto do NRF2 na desregulação da UPR em Doenças Neurodegenerativas
O mau funcionamento das enzimas PDI e a ativação crônica da RUP podem subsequentemente iniciar ou acelerar a neurodegeneração. Neurónios afectados pela doença, modelos animais de doença neurodegenerativa, bem como tecidos humanos post mortem evidenciaram a regulação positiva de vários marcadores UPR na maioria destes distúrbios. A alteração da via PDI / UPR em doenças neurodegenerativas foi bem revisada em [63], mas os seguintes destaques de amostras post-mortem no cérebro devem ser considerados. Os níveis de PDI estão aumentados em neurônios portadores de embaraços e em corpos de Lewy de pacientes com DA e DP, respectivamente [64], [65]. PDI e ERP57 são regulados positivamente no CSF em pacientes com ELA e em cérebros de indivíduos com CJD [66], [67], [68]. BIP, PERK, IRE1 e ATF6 estão elevados em amostras de pacientes com AD, PD ou ALS [69], [70], [71], [67]. BIP, CHOP e XBP1 estão elevados em amostras cerebrais post-mortem de HD [72], [73]. Além disso, a regulação positiva de ERP57, GRP94 e BIP foi encontrada em tecidos do córtex de pacientes com DCJ [74]. Em conjunto, esta evidência revela que o acúmulo de proteínas mal dobradas no parênquima cerebral leva a uma ativação deletéria e crônica da RUP. Curiosamente, há um estudo recente ligando a ativação de NRF2 por PERK no início da DA. Neste estudo, os autores analisaram se alterações mediadas por estresse oxidativo em NRF2 e UPR podem constituir eventos precoces na patogênese da DA utilizando células do sangue periférico humano e um modelo de camundongo transgênico com DA em diferentes estágios da doença. O aumento do estresse oxidativo e aumento de pSer40-NRF2 foram observados em células mononucleares do sangue periférico humano isoladas de indivíduos com comprometimento cognitivo leve. Além disso, eles relataram homeostase de cálcio do ER prejudicada e marcadores de estresse de ER regulados positivamente nessas células de indivíduos com comprometimento cognitivo leve e leve AD [75].
Regulação mútua de NRF2 e o Ubiquitin Proteasome System (UPS)
O no-break modula os níveis de proteína NRF2
O UPS participa da degradação de proteínas danificadas ou mal dobradas e controla os níveis das principais moléculas reguladoras no citosol e no núcleo. O núcleo central deste sistema é uma grande enzima com múltiplas subunidades que contém um complexo ativo proteolítico denominado 20S. O proteassoma do núcleo 20S degrada proteínas desdobradas, mas a ligação a diferentes complexos proteicos reguladores altera sua especificidade e atividade de substrato. Por exemplo, a adição de uma ou duas subunidades reguladoras 19S ao núcleo 20S constitui o proteassoma 26S e altera sua especificidade em relação às proteínas dobradas nativas [76], [77]. A degradação proteasomal necessita de ligação covalente da ubiquitina. A conjugação da ubiquitina prossegue através de um mecanismo em cascata de três etapas. Primeiro, a enzima ativadora de ubiquitina E1 ativa a ubiquitina em uma reação que requer ATP. Então, uma enzima E2 (proteína transportadora de ubiquitina ou enzima conjugadora de ubiquitina) transfere a ubiquitina ativada de E1 para o substrato que é especificamente ligado a um membro da família da ubiquitina-proteína ligase, denominada E3. Embora o destino exato da proteína ubiquitinada dependa da natureza da cadeia da ubiquitina, esse processo geralmente resulta na degradação pelo proteossomo 26S [78].
O E3-ligase KEAP1 é o inibidor mais conhecido do NRF2. O mecanismo do regulamento KEAP1 explica com elegância como os níveis de NRF2 se ajustam às flutuações do oxidante. Sob condições basais, o NRF2 recém-sintetizado é capturado pelo homodímero KEAP1, que se liga a uma molécula NRF2 em duas seqüências de aminoácidos com baixa (aspartato, leucina, glicina; DLG) e alta (glutamato, treonina, glicina, glutamato; ETGE). A interação com KEAP1 ajuda a apresentar NRF2 ao complexo de proteína CULLIN3 / RBX1, resultando em sua ubiquitinação e subsequente degradação proteasomal. No entanto, a modificação redox de KEAP1 impede a apresentação de NRF2 ao no-break representado por CULLIN3 / RBX1. Como resultado, o NRF2 recém-sintetizado escapa da degradação dependente de KEAP1, acumula-se no núcleo e ativa os genes contendo ARE [79], [80], [81], [82].
O adaptador de E3-ligase? -TrCP também é um homodímero que participa dos eventos de sinalização relacionados à fosforilação de NRF2 por GSK-3 ?. Esta quinase fosforila resíduos de serina específicos de NRF2 (aspartato, serina, glicina, isoleucina serina; DSGIS) para criar um domínio de degradação que é então reconhecido por? -TrCP e marcado para degradação de proteassoma por um complexo CULLIN1 / RBX1. A identificação dos aminoácidos específicos que são fosforilados por GSK-3? neste degron foi conduzido por uma combinação de mutagênese dirigida ao local do domínio Neh6, eletroforese em gel 2D [15], [26] e espectroscopia de massa [83]. Consequentemente, a inibição de GSK-3? por drogas altamente seletivas ou siRNAs contra isoformas GSK-3 resultaram em um aumento nos níveis de proteína NRF2. Resultados semelhantes foram encontrados com siRNAs contra as isoformas 1 e 2 de? -TrCP. Estabilizao de NRF2 seguindo GSK-3? a inibição ocorreu em fibroblastos de embrião de camundongo deficientes em KEAP1 e em um mutante de deleção de NRF2 expresso ectopicamente sem os resíduos de ETGE críticos para ligação de alta afinidade a KEAP1, demonstrando ainda uma regulação independente de KEAP1.
No contexto das doenças neurodegenerativas, podemos imaginar a modulação do NRF2 pelo UPS de duas maneiras diferentes. Por um lado, o sistema KEAP1 detectaria desequilíbrio redox derivado do acúmulo de proteína mal dobrada, enquanto o eixo GSK-3 /? - TrCP atuaria como um participante ativo na transdução de sinalização alterada pela perda de proteostase (Fig. 2).
Figura 2 O no-break controla rigidamente os níveis NRF2. Em condições homeostáticas, baixos níveis de NRF2 são mantidos pela ação dos adaptadores de ligases E3 KEAP1 e? -TrCP. À esquerda, NRF2 liga-se aos domínios Kelch de um homodímero KEAP1 através de motivos de afinidade baixa (DLG) e alta (ETGE). Por meio de seu domínio BTB, KEAP1 se liga simultaneamente a um complexo CULLIN3 / RBX1, permitindo a ubiquitinação e degradação de NRF2 pelo proteassoma 26 S. Além disso, GSK-3? fosforila resíduos Ser335 e Ser338 de NRF2 para criar um domínio de degradação (DpSGIpSL) que é então reconhecido pelo adaptador de ubiquitina ligase? -TrCP e marcado para degradação de proteassoma por um complexo CULLIN3 / RBX1. Certo, após a exposição a espécies reativas de oxigênio ou resíduos Cys críticos de eletrófilos em KEAP1 são modificados, tornando KEAP1 incapaz de interagir de forma eficiente com NRF2 ou CULLIN3 / RBX1 e, então, este fator de transcrição aumenta sua meia-vida e atividade de transcrição para genes ARE. As vias de sinalização que resultam na inibição de GSK-3 ?, como a fosforilação de AKT em Ser9, resultam na degradação prejudicada de NRF2 pelo proteassoma, acumulação e indução de genes alvo.
NRF2 aumenta a atividade da UPS através do controle transcricional das subunidades do proteassoma
O NRF2 regula positivamente a expressão de várias subunidades do proteassoma, protegendo assim a célula do acúmulo de proteínas tóxicas. Vinte genes relacionados a proteassoma e ubiquitinação parecem ser regulados por NRF2, de acordo com uma ampla análise de microarranjos de RNA de fígado que foi criado com o indutor D2T de NRF3 [84]. Em estudo posterior, os mesmos autores evidenciaram que a expressão da maioria das subunidades do proteassoma 26S aumentou em até três vezes em fígados de camundongos tratados com D3T. Os níveis de proteína da subunidade e a atividade do proteassoma aumentaram coordenadamente. No entanto, nenhuma indução foi observada em camundongos onde o fator de transcrição NRF2 foi interrompido. A atividade do promotor da subunidade do proteassoma PSMB5 (20S) aumentou com a superexpressão de NRF2 ou tratamento com ativadores em fibroblastos embrionários de camundongo, e as AREs foram identificadas no promotor proximal de PSMB5 [85]. A ativação farmacológica de NRF2 resultou em níveis elevados de expressão de subunidades de proteassoma representativas (PSMA3, PSMA6, PSMB1 e PSMB5) apenas em fibroblastos humanos não senescentes contendo NRF2 funcional [86]. A ativação de NRF2 durante a adaptação ao estresse oxidativo resulta em alta expressão de PSMB1 (20S) e PA28? subunidades (ou S11, regulador de proteassoma) [87]. Além disso, resultados de células-tronco embrionárias humanas revelaram que NRF2 controla a expressão da proteína de maturação do proteassoma (POMP), uma chaperona proteassoma, que por sua vez modula a proliferação de células-tronco embrionárias humanas que se auto-renovam, diferenciação de três camadas germinativas e reprogramação celular [ 88]. Juntos, esses estudos indicam que o NRF2 regula positivamente a expressão de componentes-chave do UPS e, portanto, contribui ativamente para a eliminação de proteínas que, de outra forma, seriam tóxicas.
O eixo NRF2-UPS em doenças neurodegenerativas
O papel do nobreak nas doenças neurodegenerativas é um campo de intenso debate. Estudos iniciais relataram diminuição da atividade do proteassoma em necropsias humanas de pacientes afetados por várias doenças neurodegenerativas. No entanto, outros estudos empregando abordagens in vitro e in vivo encontraram atividade de proteassoma inalterada ou mesmo aumentada (revisada em [89]). Uma possível explicação para essa discrepância é que os níveis dos componentes do UPS podem mudar durante a progressão da doença e em diferentes regiões cerebrais, como tem sido sugerido para os alvos NRF2.
Apesar desta controvérsia, deve-se notar que a regulação para cima de genes de proteassoma contendo ARE reforçará o UPS aumentando a depuração de proteínas tóxicas no cérebro. De fato, a ablação de NRF1, também modulador da resposta antioxidante, nas células neuronais leva a uma atividade de proteassoma prejudicada e neurodegeneração. Experimentos de imunoprecipitação cromatínica e análise transcricional demonstraram que PSMB6 é regulado por NRF1. Além disso, o perfil de expressão gênica levou à identificação de NRF1 como um regulador chave de transcrição de genes de proteassoma em neurônios, sugerindo que perturbações em NRF1 podem contribuir para a patogênese de doenças neurodegenerativas [90]. Curiosamente, o NRF1 e sua longa isoforma chamada TCF11 mostraram regular positivamente os genes de proteassoma contendo ARE após a inibição do proteassoma em um loop de feedback para compensar a atividade proteolítica reduzida [91], [92].
Em relação ao NRF2, existe uma correlação entre a redução dos níveis de NRF2, RPT6 (19 S) e PSMB5 (20 S) no mesencéfalo de camundongos deficientes em DJ-1 tratados com a neurotoxina paraquat [93]. Além disso, o sulforafano composto natural (SFN) dá uma imagem mais robusta de NRF2 como um modulador crucial do UPS. Expericias in vitro com neuroblastoma murino de culas Neuro2A evidenciaram uma express aumentada das subunidades catalicas do proteassoma, assim como as suas actividades de peptidase em resposta ao SFN. Este fármaco protegeu as células da citotoxicidade mediada pelo peróxido de hidrogénio e oxidação proteica de uma maneira dependente da função proteassoma [94]. Além disso, Liu e colegas de trabalho usaram um mouse repórter para monitorar a atividade da UPS em resposta ao SFN no cérebro. Esses camundongos exprimem de forma ubíqua a proteína de fluorescência verde (GFP) fundida a um sinal de degradação constitutivo que promove sua rápida degradação pelo UPS (GFPu). No córtex cerebral, o SFN reduziu o nível de GFPu com um aumento paralelo nas atividades semelhantes à quimotripsina (PSMB5), semelhante à caspase (PSMB2) e semelhante à tripsina (PSMB1) do proteassoma 20 S. Além disso, o tratamento de células derivadas de Huntington com SFN revelou que a ativação de NRF2 aumentou a degradação mHtt e reduziu a citotoxicidade mHtt [95]. O principal mecanismo de ação da SFN é através da indução de NRF2 [96]. A contribuição específica de NRF2 deve ser abordada empregando sistemas nulos NRF2 em estudos posteriores.
Conexão funcional entre NRF2 e macroautofagia
Níveis de Proteína NRF2 são Modulados pela Proteína Adaptadora P62
Autofagia refere-se à degradação de componentes citosólicos dentro dos lisossomos. Este processo é utilizado para a limpeza de proteínas de vida longa e com dobras incorretas, bem como organelas danificadas. Uma ligação direta entre NRF2 e autofagia foi observada pela primeira vez em conexão com a proteína adaptadora p62, também denominada SQSTM1 [97], [98], [99], [100], [101]. Esta proteína transporta proteínas ubiquitinadas para as máquinas de degradação proteossómica e lisossómica e sequestra as proteínas danificadas em agregados antes da sua degradação. O P62 apresenta um domínio associado à ubiquitina (UBA), para ligação a proteínas ubiquitinadas, e uma região de interação LC3 (LIR) para integração com a membrana autofagomática através do receptor de autofagia LC3.
Embora a indução mediada por p62 de NRF2 e seus genes alvo tenha sido relatada pela primeira vez em 2007 [102], o mecanismo molecular não foi totalmente compreendido até a descoberta de sua interação com KEAP1 [103], [98], [99], [100 ], [101]. Komatsu e colaboradores identificaram uma região de interação de KEAP1 (KIR) em p62 que se ligava a KEAP1 na mesma bolsa de superfície básica que NRF2 e com uma afinidade de ligação semelhante ao motivo ETGE em NRF2, sugerindo competição entre p62 e NRF2. A fosforilação de Ser351 no motivo KIR em p62 (349-DPSTGE-354) mostrou aumentar sua afinidade para KEAP1, competindo com a ligação de NRF2 e permitindo seu acúmulo e ativação transcricional de seus genes alvo [98], [99]. Na verdade, a superexpressão de p62 levou à redução da ubiquitinação de NRF2 e à consequente estabilização, bem como à indução de seus genes-alvo [104]. Foi sugerido que algumas quinases participam da fosforilação de p62. O alvo de mamífero do complexo 1 de rapamicina (mTORC1) pode estar implicado, uma vez que o tratamento com o inibidor de mTOR rapamicina suprimiu a fosforilação de p62 e a regulação negativa de KEAP1 após tratamento com arsenito. Recentemente, foi demonstrado que a quinase 1 ativada por TGF -? - (TAK1) também pode fosforilar p62, aumentando a degradação de KEAP1 e a regulação positiva de NRF2. Os autores deste estudo sugerem que esta é uma maneira de regular a redoxtase celular em condições de estado estacionário, uma vez que a deficiência de TAK1 regula ROS na ausência de qualquer oxidante exógeno em diferentes tecidos de camundongo em paralelo com uma redução nos níveis de proteína NRF2 [105 ]
Uma construção p62 sem o domínio UBA ainda era capaz de ligar o KEAP1, implicando que a interação não dependia do KEAP1 [101] ubiquitinado. No entanto, o homólogo p62 em Drosophila melanogaster, chamado Ref (2), não contém um motivo KIR e não interage diretamente com DmKEAP1, embora possa se ligar a DmKEAP1 ubiquitinado através do domínio UBA. Além disso, DmKEAP1 pode interagir diretamente com Atg8 (homólogo para mamíferos LC3). A deficiência de KEAP1 resulta em indução de Atg8 e autofagia dependente do NnFxNumX ortólogo CncC e independente de TFEB / MITF [2]. A relação entre NRF106 e autofagia parece ser conservada, destacando sua relevância funcional.
A indução de NRF2 por p62 é o resultado da competição para ligar KEAP1 e degradação de KEAP1 no lisossomo. O silenciamento de p62 com siRNA dobrou a meia-vida de KEAP1 em paralelo com uma diminuição em NRF2 e seus genes-alvo [101]. De acordo, a ablação da expressão de p62 evidenciou níveis aumentados de KEAP1 em comparação com ratos do tipo selvagem. Muito relevante, o incremento nos níveis de KEAP1 não foi afetado pelos inibidores de proteassomas, mas foi reduzido sob autofagia indutora de fome [107]. De fato, o KEAP1 está presente em células de mamíferos em vesículas autofágicas decoradas com p62 e LC3 [99], [100], [103]. Todos esses dados sugerem que o KEAP1 é um substrato da maquinaria de macroautofagia, mas essa questão deve ser analisada com mais detalhes, devido à existência de alguns resultados controversos. Os níveis de proteína KEAP1 foram aumentados em camundongos Atg7-nulo, um dos principais efetores da macroautofagia [107], mas a inibição farmacológica da macroautofagia com torin1, E64 / pepstatina ou bafilomicina não acumulou KEAP1 [107], [100]. No geral, estes resultados sugerem que níveis aumentados de p62 sequestram KEAP1 em vacúolos autofágicos e provavelmente estes resultados na degradação autofágica de KEAP1 permitindo a ativação de NRF2 (Fig. 3). Dois estudos diferentes relataram que as redutases de ácido sulfínico SESTRINS desempenham um papel importante neste contexto. SESTRIN 2 interage com p62, KEAP1 e RBX1 e facilita a degradação dependente de p62 da ativação de KEAP1 e NRF2 de genes alvo [108]. Outro estudo mostrou que o SESTRIN 2 interagiu com o ULK1 e o p62, promovendo a fosforilação do p62 no Ser403, o que facilitou a degradação de proteínas de carga, incluindo o KEAP1 [109].
Figura 3 NRF2 níveis são regulados pela proteína adaptadora p62. A fosforilação de Ser 351 no motivo KIR de p62 (349-DPSTGE-354) por mTORC1, TAK1 ou outras quinases resulta numa afinidade aumentada para a ligação a KEAP1 devido à semelhança com o motivo ETGE em NRF2. Como consequência, o p62 fosforilado desloca o NRF2 e liga o KEAP1. O motivo LIR no p62 permite a interação com LC3 na membrana autofagomática, de modo que o complexo p62-KEAP1 é eventualmente degradado no lisossomo. Como consequência, o NRF2 é capaz de se acumular, translocar para o núcleo e aumentar a transcrição de genes contendo ARE, incluindo o p62. Este mecanismo regulador fornece uma resposta NRF2 perdurável, pois o KEAP1 precisa ser sintetizado novamente para inibir a atividade NRF2.
Modulação de Genes de Macroautofagia por NRF2
O NRF2 regula a expressão de genes relevantes para a macroautofagia, bem como para o UPR e o UPS. A primeira evidência veio de estudos em que a expressão de p62 mostrou ser induzida após a exposição a eletrófilos, ROS e óxido nítrico [110], [111], [112]. O mecanismo de indução foi descrito alguns anos mais tarde com a descoberta de que o p62 contém um ERA funcional em seu promotor de gene [99]. Em um estudo recente, vários outros AREs funcionais foram encontrados e validados após análises de bioinformática e ensaios ChIP. Além disso, fibroblastos embrionários de camundongos e neurônios corticais de camundongos nocaute para Nrf2 exibiram expressão de p62 reduzida, que poderia ser resgatada com um lentivírus expressando NRF2. Da mesma forma, a deficiência de NRF2 reduziu os níveis de p62 em neurônios lesados do hipocampo de camundongos [36]. Portanto, foi sugerido que a ativação de NRF2 aumenta os níveis de p62, resultando na degradação de KEAP1 e favorecendo a estabilização de NRF2 em um ciclo de feedback positivo. Este mecanismo não canônico de indução de NRF2 requer mudanças na expressão gênica e pode ser uma resposta relevante ao estresse celular prolongado.
A proteína de reconhecimento de carga NDP52 mostrou ser transcricionalmente regulada por NRF2. O NDP52 funciona de forma semelhante ao p62, reconhecendo proteínas ubiquitinadas e interagindo com LC3 através de um domínio LIR, de modo que as cargas são degradadas nos lisossomos. Cinco AREs putativas foram encontradas na sequência de DNA do promotor Ndp52. Três deles foram identificados com diferentes construções mutantes e testes ChIP como indispensáveis para a transcrição Ndp2 mediada por NRF52 [113]. De notar, os níveis de ARNm de Ndp52 foram reduzidos no hipocampo de ratinhos Nrf2-knockout. Uma dessas sequências também foi validada em um estudo independente como uma ARE regulada por NRF2 [36].
No entanto, o papel do NRF2 na modulação da autofagia não se limita à indução dessas duas proteínas de reconhecimento de carga. A fim de obter uma visão mais profunda do papel da NRF2 na modulação de genes relacionados à autofagia adicionais, nosso grupo testou o banco de dados de imunoprecipitação da cromatina ENCODE para duas proteínas, MAFK e BACH1, que se ligam às AREs reguladas por NRF2. Usando um script gerado a partir da sequência de consenso ARE da JASPAR, identificamos várias AREs putativas em muitos genes de autofagia. Doze dessas sequências foram validadas como AREs reguladas por NRF2 em nove genes de autofagia, cuja expressão foi diminuída em fibroblastos de camundongos embrionários de camundongos Nrf2-knockout, mas pôde ser restaurada por um lentivírus expressando NRF2. Nosso estudo demonstrou que NRF2 ativa a expressão de alguns genes envolvidos em diferentes etapas do processo autofágico, incluindo início de autofagia (ULK1), reconhecimento de carga (p62 e NDP52), formação de autofagossomos (ATG4D, ATG7 e GABARAPL1), alongamento (ATG2B e ATG5 ) e depuração do autolisossomo (ATG4D). Consequentemente, o fluxo de autofagia em resposta ao peróxido de hidrogênio foi prejudicado quando o NRF2 estava ausente [36].
Relevância da expressão de genes de macroautofagia mediada por NRF2 em desordens neurodegenerativas
A autofagia defeituosa mostrou desempenhar um papel importante em várias doenças neurodegenerativas [114] e a ablação da autofagia leva à neurodegeneração em camundongos [115], [116]. Camundongos knockout Atg7 revelaram que a deficiência de autofagia resulta em acúmulo de p62 em corpos de inclusão positivos à ubiquitina. O KEAP1 foi sequestrado nestes corpos de inclusão, levando à estabilização do NRF2 e indução de genes alvo [103]. Importante, o acúmulo excessivo de p62 juntamente com proteínas ubiquitinated foi identificado em doenças neurodegenerativas, incluindo AD, PD e ALS [117]. De fato, neurônios expressando altos níveis de APP ou TAU de pacientes com DA também expressaram p62 e NRF2 nuclear, sugerindo sua tentativa de degradar agregados intraneuronais através da autofagia [36].
A deficiência de NRF2 agrava a agregação de proteínas no contexto da DA. Na verdade, níveis aumentados de TAU fosforilada e insolúvel em sarcosil são encontrados em camundongos nocaute para Nrf2, embora nenhuma diferença nas atividades de quinase ou fosfatase possa ser detectada em comparação com o fundo do tipo selvagem [113]. É importante ressaltar que NDP52 foi demonstrado para co-localizar com TAU em neurônios murinos e interação direta entre fosfo-TAU e NDP52 foi mostrado por experimentos de co-imunoprecipitação em camundongos e amostras de AD, apontando para seu papel na degradação TAU. Curiosamente, o silenciamento de NDP52, p62 ou NRF2 em neurônios resultou em aumento de fosfo-TAU [113], [118]. Além disso, agregados de APP intraneuronais aumentados foram encontrados no hipocampo de camundongos APP / PS1? E9 quando NRF2 estava ausente. Isso se correlacionou com marcadores de autofagia alterados, incluindo aumento das razões fosfo-mTOR / mTOR e fosfo-p70S6k / p70S6k (indicativo de inibição da autofagia), níveis aumentados de pré-catepsina D e um maior número de corpos multivesiculares [119]. Em camundongos que co-expressam APP humana (V717I) e TAU (P301L), a deficiência de NRF2 levou a níveis aumentados de fosfo-TAU total e na fração insolúvel e agregados de APP intraneuronais aumentados, juntamente com níveis neuronais reduzidos de p62, NDP52, ULK1, ATG5 e GABARAPL1. A co-localização entre a proteína adaptadora p62 e APP ou TAU foi reduzida na ausência de NRF2 [36]. No geral, esses resultados destacam a importância do NRF2 na autofagia neuronal.
Fatores diferentes de transcrição agem coordenadamente para modular a proteinase
Sob condies de estado estacionio, a proteostase controlada atrav de interaces protea-protea e modificaes p-traduo, obtendo uma resposta rida. No entanto, a adaptação celular requer a regulação transcricional dos genes UPR, UPS e autofagia. Considerando que as células nervosas são continuamente submetidas a insultos tóxicos de baixo grau, incluindo estresse oxidativo e proteotóxico, um reforço da proteostase induzida pela modulação transcricional pode ajudar a prevenir a degeneração cerebral.
No caso da UPR, a ativação de cada um dos três braços finalmente resultará na indução transcricional de certos genes (revisto em [43]). Por exemplo, um fragmento derivado de ATF6 (ATF6f) se liga a elementos de resposta a estresse ER (ERSE) e induz a expressão de vários genes, incluindo XBPI, BIP e CHOP. Além disso, a sinalização PERK leva à ativação do fator de transcrição ATF4, que controla a expressão de múltiplos genes relacionados à UPR e alguns outros, incluindo os genes alvo NRF2 Hmox1 e p62. Finalmente, a ativação de IRE1 resulta na geração de um fator de transcrição ativo, XBP1 (XBP1s), que controla a transcrição de genes que codificam proteínas envolvidas no enrolamento de proteínas.
Por outro lado, o NRF1 mostrou ser necessário para a expressão gênica proteossômica no cérebro, pois camundongos Nrf1-knockout exibiram expressão reduzida de genes codificadores de várias subunidades do núcleo 20S, bem como o complexo regulador 19S juntamente com função proteassósmica prejudicada [90 ]. Tanto o NRF1 quanto o NRF2 se ligam a seqüências de AREs nas regiões promotoras de seus genes-alvo, o que sugere que eles têm atividades transcricionais sobrepostas, apesar de diferirem em seus mecanismos regulatórios e localização celular [120].
Fatores de transcrição da família FOXO controlam a expressão de múltiplos genes relacionados à autofagia. Semelhante ao que ocorre com NRF2, existem múltiplas camadas de regulação da atividade dos membros da FOXO, que podem ser induzidas sob estresse nutricional ou oxidativo [121]. Finalmente, o fator de transcrição TFEB, considerado o principal regulador da biogênese lisossomal, desempenha um papel crucial na regulação da autofagia sob condições de estresse nutricional. Assim, a inibição de mTORC1 leva à translocação nuclear de TFEB e à indução da expressão de genes de autofagia [122].
No geral, a existência de diferentes reguladores transcricionais dessas máquinas também sugere mecanismos de crosstalk e parcialmente redundantes que podem assegurar a proteostase sob diferentes circunstâncias. Consequentemente, o NRF2 pode ter um papel relevante em tecidos que suportam altos níveis de estresse oxidativo. Por exemplo, NRF2 induzido por estresse oxidativo pode funcionar sob condições ricas em nutrientes para regular transcricionalmente a autofagia, semelhante ao que foi encontrado para TFEB sob condições de fome. Além disso, o cérebro funciona em grande parte sob condições ricas em nutrientes, colocando o NRF2 como um mecanismo relevante para ativar a autofagia nos neurônios.
Potencial terapêutico promissor para NRF2 em proteinopatias
Nos últimos anos, um grande progresso foi alcançado no conhecimento das funções regulatórias da UPR, UPS e autofagia na atividade NRF2, bem como na recíproca transcrição mediada por NRF2 dos componentes desses três sistemas. Portanto, novas possibilidades terapêuticas podem surgir com base na exploração de NRF2 como um regulador crucial da eliminação de proteínas em doenças neurodegenerativas.
No entanto, uma questão chave remanescente é se será útil ou prejudicial aumentar os níveis de NRF2 no cérebro. A análise de dados epidemiológicos pode fornecer uma resposta parcial, pois indica que o gene NFE2L2 é altamente polimórfico e alguns polimorfismos de nucleotídeo único encontrados em sua região reguladora do promotor podem fornecer uma gama de variabilidade fisiológica na expressão gênica em nível de população e alguns haplótipos foram associados com diminuição do risco e / ou início tardio de AD, PD ou ALS [123]. Além disso, conforme discutido por Hayes e colegas [124], o efeito de NRF2 pode ter uma resposta em forma de U, o que significa que níveis muito baixos de NRF2 podem resultar em uma perda de citoproteção e aumento da suscetibilidade a estressores, enquanto NRF2 em excesso pode perturbar o equilíbrio homeostático em direção um cenário redutor (estresse redutivo), que favoreceria o mal dobramento e a agregação das proteínas. Níveis baixos de NRF2 no cérebro sustentam a ideia de que uma ligeira regulação para cima pode ser suficiente para alcançar um benefício em condições patológicas. Na verdade, o papel protetor da ativação farmacológica da depuração de proteínas mediada por NRF2 foi demonstrado em diferentes culturas de células de neurodegeneração e modelos in vivo.
SFN é um ativador NRF2 farmacológico que demonstrou induzir a expressão do gene proteassomal e autofagia [95], [36]. Curiosamente, Jo e colegas demonstraram que SFN reduziu os níveis de TAU fosforilado e aumentou Beclin-1 e LC3-II, sugerindo que a ativação de NRF2 pode facilitar a degradação desta proteína tóxica por meio da autofagia [113]. Além disso, a degradação de mHtt foi aumentada com SFN, e isso foi revertido com o uso de MG132, indicando degradação proteassomal desta proteína tóxica [95]. A degradação mediada por autofagia de TAU-fosfo e insolúvel foi relatada com o flavonóide orgânico fisetina. Este composto foi capaz de induzir autofagia ao promover simultaneamente a ativação e a translocação nuclear de TFEB e NRF2, juntamente com alguns de seus genes alvo. Esta resposta foi evitada pelo silenciamento TFEB ou NRF2 [125]. Bott e colegas relataram efeitos benéficos de um ativador NRF2, NRF1 e HSF1 simultâneo na toxicidade da proteína na atrofia muscular espinhal e bulbar, um distúrbio neurodegenerativo causado pela expansão de repetições CAG que codificam poliglutamina nas quais agregados de proteínas estão presentes [126]. O potencial de ativação de NRF2 para o tratamento de doenças neurodegenerativas foi demonstrado com a aprovação de BG-12, a formulação oral do indutor de NRF2 fumarato de dimetila (DMF), para o tratamento de esclerose múltipla [127], [128]. O sucesso do DMF com doenças autoimunes com forte componente inflamatório sugere que doenças neurodegenerativas podem se beneficiar do reposicionamento desse medicamento. Em um estudo pré-clínico recente de um modelo de? -Sinucleinopatia de DP, o DMF demonstrou ser neuroprotetor devido, em parte, à sua indução de autofagia [129]. Os estudos que relatam os efeitos benéficos do NRF2 na neurodegeneração, mas não se concentram em seu efeito na depuração de proteínas, são ainda mais abundantes (para uma revisão abrangente, consulte [7]). Isso é bastante relevante, pois destaca os múltiplos processos de dano que podem ser visados simultaneamente por um único acerto no NRF2, incluindo também estresse oxidativo, neuroinflamação ou disfunção mitocondrial. No entanto, trabalhos futuros serão necessários para determinar definitivamente se a ativação farmacológica de NRF2 pode ser uma estratégia válida para facilitar a degradação de proteínas tóxicas no cérebro.
Como explicado antes, o GSK-3 exacerbado? atividade foi relatada em doenças neurodegenerativas e tem sido especulado que a redução conseqüente de NRF2 pode ser parcialmente responsável pelo resultado deletério. Nessas condições patológicas, os inibidores de GSK-3 também podem cooperar para aumentar os níveis de NRF2 e a proteostase. Os efeitos benéficos dos inibidores de GSK-3 foram relatados em diferentes modelos de neurodegeneração e, mais interessante, a repressão de GSK-3 mostrou reduzir os níveis de proteínas tóxicas [130], [131], [132], [133]. Embora nenhuma ligação direta entre a inibição de GSK-3 e a regulação da transcrição de NRF2 de genes que promovem a proteostase tenha sido observada ainda, é razoável especular que a regulação negativa da atividade de GSK-3 resultaria em níveis aumentados de NRF2, o que eventualmente resultará em reforço proteostase.
A atividade transcricional do NRF2, bem como a capacidade celular de manter a proteostase, diminuem com a idade, principal fator de risco para o desenvolvimento de doenças neurodegenerativas. É razoável pensar que o reforço do NRF2 e, conseqüentemente, a proteostase, pelo menos, retardaria o acúmulo de agregados proteicos e a neurodegeneração. De fato, o tratamento de fibroblastos senescentes humanos com ácido 18? -Glicirretínico (18? -GA) triterpenóide promoveu a ativação de NRF2, levando à indução de proteassoma e aumentando a expectativa de vida. Este estudo sugere que a ativação farmacológica de NRF2 é possível mesmo na idade avançada [86]. Além disso, um estudo posterior mostrou que este composto media a ativação de SKN-1 e proteassoma em C.elegans com efeitos benéficos na progressão da DA em modelos de nematóides relevantes [134].
Considerando tudo, a indução mediada por NRF2 de genes relacionados à proteostase parece ser benéfica em diferentes proteinopatias.
Sulforafano e seus efeitos sobre câncer, mortalidade, envelhecimento, cérebro e comportamento, doenças cardíacas e muito mais
Os isotiocianatos são alguns dos compostos vegetais mais importantes que você pode obter em sua dieta. Neste vídeo eu faço o caso mais abrangente para eles que já foi feito. Curto período de atenção? Pule para o seu tópico favorito clicando em um dos pontos de tempo abaixo. Cronograma completo abaixo.
Seções principais:
00: 01: 14 - Câncer e mortalidade
00: 19: 04 - envelhecimento
00: 26: 30 - Cérebro e comportamento
00: 38: 06 - recapitulação final
00: 40: 27 - dose
Cronograma completo:
00: 00: 34 - Introdução do sulforafano, um dos principais focos do vídeo.
00: 01: 14 - Consumo de vegetais crucíferos e reduções na mortalidade por todas as causas.
00: 02: 12 - risco de câncer de próstata.
00: 02: 23 - risco de câncer de bexiga.
00: 02: 34 - Câncer de pulmão em risco de fumantes.
00: 02: 48 - risco de câncer de mama.
00: 03: 13 - Hipotético: e se você já tem câncer? (intervencionista)
00: 03: 35 - Mecanismo plausível que direciona os dados associativos de câncer e mortalidade.
00: 04: 38 - Sulforafano e câncer.
00: 05: 32 - Evidência animal mostrando forte efeito do extrato de brócolis no desenvolvimento do tumor de bexiga em ratos.
00: 06: 06 - Efeito da suplementação direta de sulforafano em pacientes com câncer de próstata.
00: 07: 09 - Bioacumulação de metabólitos de isotiocianato no tecido mamário atual.
00: 08: 32 - Inibição de células estaminais de cancro da mama.
00: 08: 53 - Lição de História: os brassicas foram estabelecidos como tendo propriedades de saúde mesmo na Roma antiga.
00: 09: 16 - A capacidade do Sulforaphane de aumentar a excreção de carcinógeno (benzeno, acroleína).
00: 09: 51 - NRF2 como um interruptor genético através de elementos de resposta antioxidante.
00: 10: 10 - Como a ativação de NRF2 aumenta a excreção de carcinógenos via conjugados de glutationa-S.
00: 10: 34 - As couves-de-bruxelas aumentam a glutationa-S-transferase e reduzem os danos no DNA.
00: 11: 20 - Bebida de brócolis aumenta a excreção de benzeno em 61%.
00: 13: 31 - O homogenato de brócolis aumenta as enzimas antioxidantes nas vias aéreas superiores.
00: 15: 45 - Consumo de vegetais crucíferos e mortalidade por doenças cardíacas.
00: 16: 55 - Brócolis em pó melhora os lipídios no sangue e o risco geral de doenças cardíacas em diabéticos tipo 2.
00: 19: 04 - Início da seção de envelhecimento.
00: 19: 21 - dieta enriquecida com sulforafano aumenta a vida útil de besouros de 15 a 30% (em certas condições).
00: 20: 34 - Importância da baixa inflamação para a longevidade.
00: 22: 05 - Os vegetais crucíferos e o pó de brócolis parecem reduzir uma grande variedade de marcadores inflamatórios em humanos.
00: 23: 40 - Recapitulação de vídeo intermediário: câncer, seções de envelhecimento
00: 24: 14 - Estudos com ratos sugerem que o sulforafano pode melhorar a função imunológica adaptativa na velhice.
00: 25: 18 - Sulforaphane melhorou o crescimento do cabelo em um modelo de rato de calvície. Imagem no 00: 26: 10.
00: 26: 30 - Início da seção do cérebro e comportamento.
00: 27: 18 - Efeito do extrato de brócolis no autismo.
00: 27: 48 - Efeito da glucorafanina na esquizofrenia.
00: 28: 17 - Início da discussão sobre depressão (mecanismo plausível e estudos).
00: 31: Estudo 21 - Mouse usando 10 diferentes modelos de depressão induzida por estresse mostram sulforafano igualmente eficaz como fluoxetina (prozac).
00: 32: 00 - Estudo mostra a ingestão direta de glucorafanina em camundongos é igualmente eficaz na prevenção da depressão do modelo de estresse de derrota social.
00: 33: 01 - Início da seção de neurodegeneração.
00: 33: 30 - Sulforafano e doença de Alzheimer.
00: 33: 44 - Sulforaphane e doença de Parkinson.
00: 33: 51 - Sulforaphane e doença de Hungtington.
00: 34: 13 - Sulforafano aumenta as proteínas de choque térmico.
00: 34: 43 - Início da seção de traumatismo cranioencefálico.
00: 35: 01 - Sulforafano injetado imediatamente após o TBI melhora a memória (estudo do mouse).
00: 35: 55 - Sulforafano e plasticidade neuronal.
00: 36: 32 - Sulforaphane melhora o aprendizado em modelos de diabetes tipo II em camundongos.
00: 37: 19 - Sulforafano e distrofia muscular de duchenne.
00: 37: 44 - Inibição da miostatina em células satélites musculares (in vitro).
00: 38: 06 - Recapitulação de vídeo tardio: mortalidade e câncer, danos no DNA, estresse oxidativo e inflamação, excreção de benzeno, doença cardiovascular, diabetes tipo II, efeitos no cérebro (depressão, autismo, esquizofrenia, neurodegeneração), via NRF2.
00: 40: 27 - Pensamentos em descobrir uma dose de brotos de brócolis ou sulforafano.
00: 41: 01 - Anedotas sobre brotar em casa.
00: 43: 14 - Nas temperaturas de cozimento e atividade de sulforafano.
00: 43: 45 - Conversão da bactéria intestinal do sulforafano da glucorafanina.
00: 44: 24 - Os suplementos funcionam melhor quando combinados com a mirosinase ativa de vegetais.
00: 44: 56 - Técnicas de cozinha e vegetais crucíferos.
00: 46: 06 - Isotiocianatos como sendo goitrogénios.
O fator 2 relacionado ao fator 2 (NF-E2) derivado do fator nuclear, também conhecido como Nrf2, é um fator de transcrição que regula a expressão de uma variedade de enzimas antioxidantes e desintoxicantes. Estudos de pesquisa também demonstraram seu papel no controle do estresse oxidativo. A maioria das doenças neurodegenerativas, como a doença de Alzheimer e a doença de Parkinson, é caracterizada por estresse oxidativo e inflamação crônica, os alvos comuns Abordagens de tratamento Nrf2. Dr. Alex Jimenez DC, Insight CCST
Observações finais
O fator de transcrição NRF2 orquestra uma resposta proteostática detectando e modulando as mudanças no UPR, UPS e autofagia (Fig. 4). Consequentemente, demonstrou-se que a falta de NRF2 agrava a proteinopatia, sugerindo que NRF2 é necessário para um clearance de proteína ótimo. Todos juntos, podemos especular que NRF2 pode ser um alvo terapêutico interessante para proteinopatias.
Figura 4 NRF2 como um hub conectando sinais de emergência derivados de proteotóxicos para uma resposta transcricional protetora. O acúmulo de proteínas desdobradas / desdobradas levará à ativação da resposta protéica desdobrada (UPR) no RE. A ativação de PERK ou MAPK pode resultar na indução transcricional do Gpx8 residente no ER e várias enzimas que regulam os níveis de GSH, essenciais para garantir o correto dobramento das proteínas. Agregados de proteínas inibem a atividade do proteassoma (UPS), provavelmente evitando a degradação do NRF2. O NRF2 demonstrou modular especificamente a transcrição dos genes Psma3, Psma6, Psmb1, Psmb5 e Pomp. Várias outras subunidades foram supra-reguladas de uma maneira dependente de NRF2 em resposta a D3T, provavelmente ampliando a lista de subunidades de proteassomo reguladas por NRF2. A autofagia é o principal caminho para a degradação de agregados proteicos. A autofagia também regula o NRF2, conectando esse caminho de degradação com a indução transcricional NRF2 de p62, Ndp52, Ulk1, Atg2b, Atg4c, Atg5, Atg7 e Gabarapl1.
De acordo com o artigo acima, enquanto os sintomas das doenças neurodegenerativas podem ser tratados através de uma variedade de opções de tratamento, estudos de pesquisa demonstraram que a ativação do Nrf2 pode ser uma abordagem de tratamento útil. Porque Os ativadores da Nrf2 visam amplos mecanismos da doença, todas as doenças neurodegenerativas podem se beneficiar do uso do fator de transcrição Nrf2. As descobertas do Nrf2 revolucionaram o tratamento de doenças neurodegenerativas. O escopo de nossas informações é limitado a questões de quiropraxia e saúde da coluna vertebral. Para discutir o assunto, sinta-se à vontade para perguntar ao Dr. Jimenez ou entre em contato conosco em 915-850-0900 .
Discussão Adicional do Tópico: Aliviar a Dor no Joelho sem Cirurgia
A dor no joelho é um sintoma bem conhecido que pode ocorrer devido a uma variedade de lesões e / ou condições no joelho, incluindo lesões esportivas. O joelho é uma das articulações mais complexas do corpo humano, pois é formado pela intersecção de quatro ossos, quatro ligamentos, vários tendões, dois meniscos e cartilagem. De acordo com a Academia Americana de Médicos de Família, as causas mais comuns de dor no joelho incluem subluxação patelar, tendinite patelar ou joelho de saltador e doença de Osgood-Schlatter. Embora a dor no joelho seja mais provável de ocorrer em pessoas com mais de 60 anos, a dor no joelho também pode ocorrer em crianças e adolescentes. Dor no joelho pode ser tratada em casa seguindo os métodos RICE; no entanto, lesões graves no joelho podem exigir atenção médica imediata, incluindo cuidados quiropráticos.
O estresse oxidativo é descrito como dano celular causado por radicais livres, ou moléculas instáveis, que podem afetar a função saudável. O corpo humano cria radicais livres para neutralizar bactérias e vírus, no entanto, fatores externos, como oxigênio, poluição e radiação, muitas vezes também podem produzir radicais livres. O estresse oxidativo tem sido associado a vários problemas de saúde.
O estresse oxidativo e outros estressores ativam mecanismos internos de proteção que podem ajudar a regular a resposta antioxidante do corpo humano. Nrf2 é uma proteína que detecta níveis de estresse oxidativo e permite que as células se protejam de fatores internos e externos. O Nrf2 também demonstrou ajudar a regular genes envolvidos na produção de enzimas antioxidantes e genes de resposta ao estresse. O objetivo do artigo abaixo é explicar a efeitos de Nrf2 no câncer.
Sumário
A via Keap1-Nrf2 é o principal regulador das respostas citoprotetoras ao estresse oxidativo e eletrofílico. Embora as vias de sinalizao celular desencadeadas pelo factor de transcrio Nrf2 previnam o inio e a progress do cancro em tecidos normais e prmalignos, em culas totalmente malignas, a actividade Nrf2 proporciona uma vantagem de crescimento aumentando a quimiorresistcia do cancro e aumentando o crescimento de culas tumorais. Nesta revisão gráfica, fornecemos uma visão geral da via Keap1-Nrf2 e sua desregulação em células cancerígenas. Também resumimos brevemente as conseqüências da ativação constitutiva de Nrf2 em células cancerosas e como isso pode ser explorado na terapia gênica do câncer.
Palavras-chave:Nrf2, Keap1, Câncer, Elemento de resposta antioxidante, Terapia gênica
Introdução
A via Keap1-Nrf2 é o principal regulador das respostas citoprotetoras a estresses endógenos e exógenos causados por espécies reativas de oxigênio (ROS) e eletrófilos [1]. As proteínas de sinalização chave dentro da via são o fator de transcrição Nrf2 (fator 2 relacionado ao eritróide 2 do fator nuclear) que se liga a pequenas proteínas Maf ao elemento de resposta antioxidante (ARE) nas regiões regulatórias dos genes alvo, e Keap1 (Kelch ECH associação da proteína 1), uma proteína repressora que se liga ao Nrf2 e promove sua degradação pela via do proteassoma da ubiquitina (Fig. 1). Keap1 é uma proteína muito rica em cisteína, Keap1 de camundongo tendo um total de 25 e humanos 27 resíduos de cisteína, muitos dos quais podem ser modificados in vitro por diferentes oxidantes e eletrófilos [2]. Três desses resíduos, C151, C273 e C288, mostraram desempenhar um papel funcional ao alterar a conformação de Keap1 levando à translocação nuclear de Nrf2 e subsequente expressão do gene alvo [3] (Fig. 1). O mecanismo exato pelo qual as modificações de cisteína em Keap1 levam à ativação de Nrf2 não é conhecido, mas os dois modelos predominantes, mas não mutuamente exclusivos, são (1) o modelo hinge e latch , no qual as modificações Keap1 em resíduos de tiol residindo no IVR de Keap1 interromper a interação com Nrf2 causando um desalinhamento dos resíduos de lisina dentro de Nrf2 que não podem mais ser poliubiquitinilados e (2) o modelo no qual a modificação do tiol causa a dissociação de Cul3 de Keap1 [3] Em ambos os modelos, o Keap2 modificado por indutor e ligado a Nrf1 é inativado e, consequentemente, as proteínas Nrf2 recentemente sintetizadas contornam Keap1 e se translocam para o núcleo, ligam-se ao ARE e conduzem a expressão de genes alvo Nrf2, como NAD (P) H quinona oxidoredutase 1 (NQO1), heme oxigenase 1 (HMOX1), glutamato-cisteína ligase (GCL) e glutationa S transferases (GSTs) (Fig. 2). Além das modificações dos tióis Keap1 resultando na indução do gene alvo Nrf2, proteínas como p21 e p62 podem se ligar a Nrf2 ou Keap1, interrompendo assim a interação entre Nrf2 e Keap1 [1], [3] (Fig. 3).
Fig. 1. Estruturas de Nrf2 e Keap1 e o código da cisteína. (A) Nrf2 consiste em 589 aminoácidos e tem seis domínios evolutivamente altamente conservados, Neh1-6. Neh1 contém um motivo bZip, uma estrutura leucine zipper (L-Zip) da região básica, onde a região básica é responsável pelo reconhecimento do DNA e o L-Zip medeia a dimerização com pequenas proteínas Maf. Neh6 funciona como um degron para mediar a degradação de Nrf2 no núcleo. Neh4 e 5 são domínios de transativação. Neh2 contém motivos ETGE e DLG, que são necessários para a interação com Keap1, e uma região hidrofílica de resíduos de lisina (7 K), que são indispensáveis para a poliubiquitinação dependente de Keap1 e degradação de Nrf2. (B) Keap1 consiste em 624 resíduos de aminoácidos e tem cinco domínios. Os dois motivos de interação proteína proteína, o domínio BTB e o domínio Kelch, são separados pela região interveniente (IVR). O domínio BTB juntamente com a porção N-terminal do IVR medeia a homodimerização de Keap1 e a ligação com Cullin3 (Cul3). O domínio Kelch e a região C-terminal medeiam a interação com Neh2. (C) Nrf2 interage com duas moléculas de Keap1 por meio de seus motivos Neh2 ETGE e DLG. Tanto o ETGE quanto o DLG se ligam a locais semelhantes na superfície inferior do motivo Keap1 Kelch. (D) Keap1 é rico em resíduos de cisteína, com 27 cisteínas em proteína humana. Algumas dessas cisteínas estão localizadas perto de resíduos básicos e, portanto, são excelentes alvos de eletrófilos e oxidantes. O padrão de modificação dos resíduos de cisteína por eletrófilos é conhecido como código de cisteína. A hipótese do código de cisteína propõe que agentes ativadores de Nrf2 estruturalmente diferentes afetam diferentes cisteínas Keap1. As modificações de cisteína levam a mudanças conformacionais no Keap1 interrompendo a interação entre os domínios Nrf2 DLG e Keap1 Kelch, inibindo assim a poliubiquitinação de Nrf2. A importância funcional de Cys151, Cys273 e Cys288 foi demonstrada, uma vez que Cys273 e Cys288 são necessários para a supressão de Nrf2 e Cys151 para ativação de Nrf2 por indutores [1], [3].
Fig. 2. O caminho de sinalização Nrf2-Keap1. (A e B) em condições basais, duas moléculas Keap1 ligam-se a Nrf2 e Nrf2 é poliubiquitilada pelo complexo E3 ligase baseado em Cul3. Essa polibiquitilação resulta em rápida degradação do Nrf2 pelo proteassoma. Uma pequena proporção de Nrf2 escapa do complexo inibitório e se acumula no núcleo para mediar a expressão gênica basal dependente de ARE, mantendo assim a homeostase celular. (C) Sob condições de estresse, os indutores modificam as cisteínas Keap1 levando à inibição da ubiquitilação de Nrf2 via dissociação do complexo inibitório. (D) De acordo com o modelo de dobradiça e trinco, a modificação de resíduos de cisteína Keap1 específicos leva a alterações conformacionais em Keap1 resultando na separação do motivo Nrf2 DLG de Keap1. A ubiquitinação de Nrf2 é interrompida, mas a ligação com o motivo ETGE permanece. (E) No modelo de dissociação Keap1-Cul3, a ligação de Keap1 e Cul3 é interrompida em resposta a eletrófilos, levando ao escape de Nrf2 do sistema de ubiquitinação. Em ambos os modelos sugeridos, o Keap2 modificado por indutor e ligado a Nrf1 é inativado e, conseqüentemente, as proteínas Nrf2 recém sintetizadas contornam Keap1 e translocam para o núcleo, ligam-se ao Elemento de Resposta Antioxidante (ARE) e direcionam a expressão do alvo Nrf2 genes tais como NQO1, HMOX1, GCL e GST [1], [3].
Fig. 3. Mecanismos para acumulação nuclear constitutiva de Nrf2 em câncer. (A) Mutações somáticas em Nrf2 ou Keap1 interrompem a interação dessas duas proteínas. Em Nrf2, as mutações afetam os motivos ETGE e DLG, mas as mutações Keap1 são distribuídas de maneira mais uniforme. Além disso, a ativação oncogênica, como KrasG12D [5], ou a interrupção de supressores de tumor, como PTEN [11], podem levar à indução transcricional de Nrf2 e a um aumento na Nrf2 nuclear. (B) A hipermetilação do promotor Keap1 no câncer de pulmão e próstata leva à redução da expressão do RNAm de Keap1, o que aumenta o acúmulo nuclear de Nrf2 [6], [7]. (C) No carcinoma renal papilar familiar, a perda da atividade da enzima fumarato hidratase leva ao acúmulo de fumarato e à succinetração de resíduos de cisteína Keap1 (2SC). Esta modificação pós-traducional leva à interrupção da interação Keap1-Nrf2 e ao acúmulo nuclear de Nrf2 [8], [9]. (D) O acúmulo de proteínas disruptoras como p62 e p21 pode perturbar a ligação de Nrf2-Keap1 e resulta em um aumento no Nrf2 nuclear. O p62 liga-se ao Keap1 sobrepondo-se ao pocket de ligação para o Nrf2 e o p21 interage diretamente com os motivos DLG e ETGE do Nrf2, competindo assim com o Keap1 [10].
Mecanismos de Ativação e Desregulação do Nrf2 no Câncer
Embora a citoproteção proporcionada pela ativação do Nrf2 seja importante para a quimioprevenção do câncer em tecidos normais e pré-malignos, em células totalmente malignas, a atividade do Nrf2 fornece vantagem de crescimento ao aumentar a quimiorresistência ao câncer e aumentar o crescimento de células tumorais [4]. Vários mecanismos pelos quais a via de sinalização Nrf2 é constitutivamente ativada em vários cânceres foram descritos: (1) mutações somáticas em Keap1 ou o domínio de ligação Keap1 de Nrf2 interrompendo sua interação; (2) silenciamento epigenético da expressão Keap1 levando a repressão defeituosa de Nrf2; (3) acúmulo de proteínas disruptoras, como p62, levando à dissociação do complexo Keap1-Nrf2; (4) indução transcricional de Nrf2 por oncogênico K-Ras, B-Raf e c-Myc; e (5) modificação pós-translacional de cisteínas Keap1 por succinilação que ocorre no carcinoma renal papilar familiar devido à perda da atividade da enzima fumarato hidratase [3], [4], [5], [6], [7], [ 8], [9], [10] (Fig. 3). A proteína Nrf2 constitutivamente abundante causa aumento na expressão de genes envolvidos no metabolismo de drogas, aumentando assim a resistência a drogas quimioterápicas e radioterapia. Além disso, níveis elevados de proteína Nrf2 estão associados a um mau prognóstico no câncer [4]. O Nrf2 hiperativo também afeta a proliferação celular, direcionando a glicose e a glutamina para vias anabólicas que aumentam a síntese de purinas e influenciam a via das pentoses fosfato para promover a proliferação celular [11] (Fig. 4).
Fig. 4. O duplo papel do Nrf2 na tumorigênese. Sob condições fisiológicas, baixos níveis de Nrf2 nuclear são suficientes para a manutenção da homeostase celular. O Nrf2 inibe a iniciação do tumor e a metástase do câncer, eliminando os carcinogênicos, as ROS e outros agentes que danificam o DNA. Durante a tumorigênese, o acúmulo de danos no DNA leva à hiperatividade constitutiva do Nrf2, que ajuda as células malignas autônomas a suportar altos níveis de EROs endógenos e evitar a apoptose. Os níveis Nrf2 nucleares persistentemente elevados ativam genes metabólicos, além dos genes citoprotetores, que contribuem para a reprogramação metabólica e aumentam a proliferação celular. Cânceres com altos níveis de Nrf2 estão associados a prognóstico ruim devido à radio e quimiorresistência e proliferação agressiva de células cancerígenas. Assim, a atividade da via Nrf2 é protetora nos estágios iniciais da tumorigênese, mas é prejudicial nos últimos estágios. Portanto, para a prevenção do câncer, a melhoria da atividade do Nrf2 continua sendo uma abordagem importante, enquanto que para o tratamento do câncer, a inibição do Nrf2 é desejável [4], [11].
Dado que a alta atividade da Nrf2 ocorre comumente em células cancerígenas com desfechos adversos, há necessidade de terapias para inibir a Nrf2. Infelizmente, devido à semelhança estrutural com alguns outros membros da família bZip, o desenvolvimento de inibidores específicos de Nrf2 é uma tarefa desafiadora e apenas alguns estudos sobre a inibição de Nrf2 foram publicados até o momento. Através da triagem de produtos naturais, Ren et al. [12] identificou um composto antineoplásico brusatol como um inibidor Nrf2 que aumenta a eficácia quimioterápica da cisplatina. Além disso, os inibidores PI3K [11], [13] e o NRF2 siRNA [14] foram utilizados para inibir o Nrf2 em células cancerígenas. Recentemente, utilizamos uma abordagem alternativa, conhecida como terapia gênica suicida de câncer, para atingir células cancerígenas com altos níveis de Nrf2. Os vetores lentivirais dirigidos por Nrf2 [15] contendo timidina quinase (TK) são transferidos para células cancerígenas com alta atividade ARE e as células são tratadas com um pró-fármaco, ganciclovir (GCV). O GCV é metabolizado em monofosfato de GCV, que é posteriormente fosforilado por quinases celulares em uma forma de trifosfato tóxico [16] (Fig. 5). Isto conduz à morte eficaz não só das células tumorais contendo TK, mas também das células vizinhas devido ao efeito espectador [17]. A terapia gênica TK / GCV regulada por ARE pode ser melhorada através da combinação de um agente quimioterápico de câncer, doxorrubicina, ao tratamento [16], apoiando a noção de que essa abordagem poderia ser útil em conjunto com terapias tradicionais.
Fig. 5. Terapia gênica suicida. O acúmulo nuclear Nrf2 constitutivo em células cancerígenas pode ser explorado usando o vetor viral Nrf2 para terapia gênica suicida de câncer [16]. Nesta abordagem, o vector lentiviral (LV) que expressa timidina cinase (TK) sob o promotor mínimo de SV40 com quatro AREs é transduzido para células de adenocarcinoma de pulmão. Níveis elevados de Nrf2 nuclear levam à expressão robusta de TK através da ligação Nrf2. As células são então tratadas com um pró-fármaco, o ganciclovir (GCV), que é fosforilado pela TK. O GCV trifosforilado interrompe a síntese de DNA e leva à morte efetiva não apenas das células tumorais contendo TK, mas também das células vizinhas devido ao efeito espectador.
Nrf2 é um regulador mestre que desencadeia a produção de poderosos antioxidantes no corpo humano que ajudam a eliminar o estresse oxidativo. Várias enzimas antioxidantes, como a superóxido dismutase, ou SOD, glutationa e catalase, também são ativadas pela via Nrf2. Além disso, certos fitoquímicos como cúrcuma, ashwagandha, bacopa, chá verde e cardo de leite ativam o Nrf2. Estudos de pesquisa descobriram que Ativação Nrf2 pode naturalmente melhorar a proteção celular e restaurar o equilíbrio do corpo humano.
Dr. Alex Jimenez DC, Insight CCST
Sulforafano e seus efeitos sobre câncer, mortalidade, envelhecimento, cérebro e comportamento, doenças cardíacas e muito mais
Os isotiocianatos são alguns dos compostos vegetais mais importantes que você pode obter em sua dieta. Neste vídeo eu faço o caso mais abrangente para eles que já foi feito. Curto período de atenção? Pule para o seu tópico favorito clicando em um dos pontos de tempo abaixo. Cronograma completo abaixo.
Seções principais:
00: 01: 14 - Câncer e mortalidade
00: 19: 04 - envelhecimento
00: 26: 30 - Cérebro e comportamento
00: 38: 06 - recapitulação final
00: 40: 27 - dose
Cronograma completo:
00: 00: 34 - Introdução do sulforafano, um dos principais focos do vídeo.
00: 01: 14 - Consumo de vegetais crucíferos e reduções na mortalidade por todas as causas.
00: 02: 12 - risco de câncer de próstata.
00: 02: 23 - risco de câncer de bexiga.
00: 02: 34 - Câncer de pulmão em risco de fumantes.
00: 02: 48 - risco de câncer de mama.
00: 03: 13 - Hipotético: e se você já tem câncer? (intervencionista)
00: 03: 35 - Mecanismo plausível que direciona os dados associativos de câncer e mortalidade.
00: 04: 38 - Sulforafano e câncer.
00: 05: 32 - Evidência animal mostrando forte efeito do extrato de brócolis no desenvolvimento do tumor de bexiga em ratos.
00: 06: 06 - Efeito da suplementação direta de sulforafano em pacientes com câncer de próstata.
00: 07: 09 - Bioacumulação de metabólitos de isotiocianato no tecido mamário atual.
00: 08: 32 - Inibição de células estaminais de cancro da mama.
00: 08: 53 - Lição de História: os brassicas foram estabelecidos como tendo propriedades de saúde mesmo na Roma antiga.
00: 09: 16 - A capacidade do Sulforaphane de aumentar a excreção de carcinógeno (benzeno, acroleína).
00: 09: 51 - NRF2 como um interruptor genético através de elementos de resposta antioxidante.
00: 10: 10 - Como a ativação de NRF2 aumenta a excreção de carcinógenos via conjugados de glutationa-S.
00: 10: 34 - As couves-de-bruxelas aumentam a glutationa-S-transferase e reduzem os danos no DNA.
00: 11: 20 - Bebida de brócolis aumenta a excreção de benzeno em 61%.
00: 13: 31 - O homogenato de brócolis aumenta as enzimas antioxidantes nas vias aéreas superiores.
00: 15: 45 - Consumo de vegetais crucíferos e mortalidade por doenças cardíacas.
00: 16: 55 - Brócolis em pó melhora os lipídios no sangue e o risco geral de doenças cardíacas em diabéticos tipo 2.
00: 19: 04 - Início da seção de envelhecimento.
00: 19: 21 - dieta enriquecida com sulforafano aumenta a vida útil de besouros de 15 a 30% (em certas condições).
00: 20: 34 - Importância da baixa inflamação para a longevidade.
00: 22: 05 - Os vegetais crucíferos e o pó de brócolis parecem reduzir uma grande variedade de marcadores inflamatórios em humanos.
00: 23: 40 - Recapitulação de vídeo intermediário: câncer, seções de envelhecimento
00: 24: 14 - Estudos com ratos sugerem que o sulforafano pode melhorar a função imunológica adaptativa na velhice.
00: 25: 18 - Sulforaphane melhorou o crescimento do cabelo em um modelo de rato de calvície. Imagem no 00: 26: 10.
00: 26: 30 - Início da seção do cérebro e comportamento.
00: 27: 18 - Efeito do extrato de brócolis no autismo.
00: 27: 48 - Efeito da glucorafanina na esquizofrenia.
00: 28: 17 - Início da discussão sobre depressão (mecanismo plausível e estudos).
00: 31: Estudo 21 - Mouse usando 10 diferentes modelos de depressão induzida por estresse mostram sulforafano igualmente eficaz como fluoxetina (prozac).
00: 32: 00 - Estudo mostra a ingestão direta de glucorafanina em camundongos é igualmente eficaz na prevenção da depressão do modelo de estresse de derrota social.
00: 33: 01 - Início da seção de neurodegeneração.
00: 33: 30 - Sulforafano e doença de Alzheimer.
00: 33: 44 - Sulforaphane e doença de Parkinson.
00: 33: 51 - Sulforaphane e doença de Hungtington.
00: 34: 13 - Sulforafano aumenta as proteínas de choque térmico.
00: 34: 43 - Início da seção de traumatismo cranioencefálico.
00: 35: 01 - Sulforafano injetado imediatamente após o TBI melhora a memória (estudo do mouse).
00: 35: 55 - Sulforafano e plasticidade neuronal.
00: 36: 32 - Sulforaphane melhora o aprendizado em modelos de diabetes tipo II em camundongos.
00: 37: 19 - Sulforafano e distrofia muscular de duchenne.
00: 37: 44 - Inibição da miostatina em células satélites musculares (in vitro).
00: 38: 06 - Recapitulação de vídeo tardio: mortalidade e câncer, danos no DNA, estresse oxidativo e inflamação, excreção de benzeno, doença cardiovascular, diabetes tipo II, efeitos no cérebro (depressão, autismo, esquizofrenia, neurodegeneração), via NRF2.
00: 40: 27 - Pensamentos em descobrir uma dose de brotos de brócolis ou sulforafano.
00: 41: 01 - Anedotas sobre brotar em casa.
00: 43: 14 - Nas temperaturas de cozimento e atividade de sulforafano.
00: 43: 45 - Conversão da bactéria intestinal do sulforafano da glucorafanina.
00: 44: 24 - Os suplementos funcionam melhor quando combinados com a mirosinase ativa de vegetais.
00: 44: 56 - Técnicas de cozinha e vegetais crucíferos.
00: 46: 06 - Isotiocianatos como sendo goitrogénios.
Agradecimentos
Este trabalho foi apoiado pela Academia da Finlândia, a Fundação Sigrid Juselius e as organizações finlandesas de câncer.
Em conclusão, o fator nuclear (derivado do eritróide 2) -like 2, também conhecido como NFE2L2 ou Nrf2, é uma proteína que aumenta a produção de antioxidantes que protegem o corpo humano do estresse oxidativo. Conforme descrito acima, a estimulação da via Nrf2 está sendo estudada para o tratamento de doenças causadas pelo estresse oxidativo, incluindo o câncer. O escopo de nossas informações é limitado a questões de quiropraxia e saúde da coluna vertebral. Para discutir o assunto, sinta-se à vontade para perguntar ao Dr. Jimenez ou entre em contato conosco em 915-850-0900 .
Discussão Adicional do Tópico: Aliviar a Dor no Joelho sem Cirurgia
A dor no joelho é um sintoma bem conhecido que pode ocorrer devido a uma variedade de lesões e / ou condições no joelho, incluindo lesões esportivas. O joelho é uma das articulações mais complexas do corpo humano, pois é formado pela intersecção de quatro ossos, quatro ligamentos, vários tendões, dois meniscos e cartilagem. De acordo com a Academia Americana de Médicos de Família, as causas mais comuns de dor no joelho incluem subluxação patelar, tendinite patelar ou joelho de saltador e doença de Osgood-Schlatter. Embora a dor no joelho seja mais provável de ocorrer em pessoas com mais de 60 anos de idade, a dor no joelho também pode ocorrer em crianças e adolescentes. A dor no joelho pode ser tratada em casa seguindo os métodos do RICE, no entanto, lesões graves no joelho podem exigir atenção médica imediata, incluindo tratamento quiroprático.
O DNA suporta aproximadamente genes 20,000, cada um segurando um programa para a criação de uma proteína ou enzima necessária para um estilo de vida saudável. Cada um desses padrões precisa ser constantemente regulado por um tipo de “promotor” que administra exatamente quanto de cada substância e / ou produto químico é gerado e sob quais condições elas também se desenvolverão.
Ao conectar-se a um tipo particular de áreas de promotor tipo switch, conhecido como o Elemento de Resposta Antioxidante, ou ARE, o Fator Nrf2 suporta a velocidade de criação de centenas de genes distintos que permitem que as células sobrevivam em circunstâncias estressantes. Esses genes, então, geram uma seleção de enzimas antioxidantes que desenvolvem uma rede de defesa neutralizando os oxidantes e limpando os subprodutos tóxicos deixados em sua produção, além de ajudar a restaurar os danos que causaram.
O que é estresse oxidativo?
Vários oxidantes, como o radical superóxido, ou O2-., E o peróxido de hidrogênio, ou H2O2, foram criados pela prática da queima de substâncias e / ou produtos químicos que sustentam o corpo humano. O corpo humano possui enzimas antioxidantes que neutralizam e desintoxicam os alimentos e bebidas reativas que consumimos. O Nrf2 modula sua produção para manter o equilíbrio e ressalta a demanda por todas essas enzimas. Esse equilíbrio pode ser interrompido por vários fatores, incluindo a idade.
À medida que envelhecemos, “o corpo humano cria menos Nrf2 e esse equilíbrio delicado pode gradualmente começar a se voltar para o lado oxidativo, um estado conhecido como estresse oxidativo. A doença também pode causar a superprodução de oxidantes. Infecções, alergias e doenças autoimunes podem, adicionalmente, acionar nossas células imunológicas para criar oxidantes reativos, como o O2-. , H2O2, OH e HOCl, onde células saudáveis são danificadas e respondem com inflamação. Doenças associadas ao envelhecimento, incluindo ataques cardíacos, derrames, câncer e condições neurodegenerativas como a doença de Alzheimer, também aumentam o desenvolvimento de oxidantes, gerando estresse e uma resposta à inflamação.
O que são ativadores Nrf2?
A proteína Nrf2, também chamada de fator de transcrição devido ao modo como ela pode suportar e controlar enzimas e genes, é o elemento secreto de uma seqüência de reações bioquímicas dentro da célula que reage a modificações no equilíbrio cognitivo e no balanço oxidativo. Os elementos sensoriais dessa via modificam e descarregam o Nrf2, acionando-o para que ele possa se espalhar no núcleo da célula em direção ao DNA. O Nrf2 pode alternativamente ligar ou desligar os genes e enzimas que ele suporta para proteger a célula.
Felizmente, uma variedade de substâncias que são ativadores Nrf2 se desenvolvem através do consumo de certas plantas e extratos utilizados séculos atrás em remédios tradicionais chineses e nativos americanos. Estes fitoquímicos parecem ser tão poderosos com menos efeitos colaterais, como os produtos farmacêuticos que ativam Nrf2 e que estão sendo usados atualmente.
Fator nuclear eritróide O fator relacionado ao 2, mais comumente conhecido como Nrf2, é um fator de transcrição que protege a célula regulando genes, enzimas e respostas antioxidantes. Fatores de transcrição são um tipo de proteína que se liga ao DNA para promover a criação de substâncias e substâncias químicas específicas, incluindo glutationa S-transferases, ou GSTs. A ativação de Nrf2 induz a produção de proteínas ativas que exibem uma poderosa capacidade antioxidante para ajudar a diminuir o estresse oxidativo.
Dr. Alex Jimenez DC, Insight CCST
A ciência por trás da ativação do Nrf2
Uma vez que o suplemento dietético Nrf2 inicial foi criado em 2004, informações mínimas foram conhecidas sobre a função da via Nrf2. Aproximadamente 200 jornais na literatura sobre Nrf2, também conhecido como nuclear factor-like 2 ou NFE2L2, existiam e os pesquisadores estavam apenas começando a descobrir a resposta antioxidante de Nrf2 em mamíferos. A partir da 2017, no entanto, mais de 9,300 pesquisas acadêmicas sobre este "regulador mestre", foram impressas.
Na realidade, o Nrf2 regula muitas enzimas antioxidantes que não se correlacionam com os genes, em vez disso, elas oferecem proteção contra uma variedade de circunstâncias relacionadas ao estresse que são encontradas pelas células, órgãos e organismos, sob condições saudáveis e patológicas. Com base nessa nova quantidade de informações de pesquisas acadêmicas publicadas, os pesquisadores agora podem desenvolver Suplementos dietéticos Nrf2.
A partir de 2007, estudos de pesquisa demonstraram a função complexa da via do Nrf2. Os ativadores Nrf2 foram encontrados para imitar fatores de diferentes estruturas dentro do corpo humano. Por meio dessas vias, os ativadores do Nrf2 foram equipados para sentir as condições de mudança em toda a célula, a fim de manter o equilíbrio e responder aos requisitos em evolução dos genes.
Por que usar suplementos de ativação Nrf2?
Como as habilidades de ativação do Nrf2 diminuem com a idade nos organismos, mudanças podem começar a ocorrer. Estudos de pesquisa demonstraram que o foco do Nrf2 em células diminui com a idade, mostrando aumento dos marcadores de estresse oxidativo. Uma variedade de doenças relacionadas à idade, como aterosclerose e doenças cardiovasculares, artrite, câncer, obesidade, diabetes tipo 2, hipertensão, catarata e doença de Alzheimer, bem como doenças de Parkinson, podem se desenvolver devido a essas mudanças. O estresse oxidativo foi encontrado com esses problemas de saúde.
Estimulando a capacidade da célula de aumentar a produção de ativadores Nrf2, Suplementos dietéticos Nrf2 pode ajudar a reviver a capacidade do próprio corpo humano de neutralizar os efeitos do estresse oxidativo. Os ácidos graxos poliinsaturados, ou PUFAs, são uma das moléculas mais prontamente oxidadas e são particularmente vulneráveis a sofrer danos causados pelos radicais livres. A produção de ácido tiobarbitúrico, ou TBARS, pode aumentar com a idade, indicando aumento do estresse oxidativo, juntamente com uma queda nas vias reguladas pelo Nrf2.
Biologicamente, a indução gênica é um mecanismo muito lento, geralmente levando horas para ser transferido por uma via. Como resultado, muitas enzimas possuem seus próprios botões liga / desliga, que podem ser acionados em minutos por diferentes enzimas regulatórias. Os pesquisadores desenvolveram composições proprietárias de ativadores Nrf2 que utilizam essa base de conhecimento de ativação. A ativação de Nrf2 é composta não apenas pelo fator de transcrição Nrf2 sendo descarregado de seu inibidor e migrando para o núcleo da célula, mas também se ligando a sequências de DNA específicas para estimular a expressão do gene citoprotetor, regulando o ritmo em que Nrf2 é retirado do núcleo.
A compreensão do procedimento de eliminação e ativação do Nrf2 no corpo humano tem permitido aos pesquisadores construir combinações de diferentes ativadores do Nrf2 para realizar a reflexão dos genes por meio de sua modulação. A combinação da base de conhecimento, juntamente com a ampla variedade de outros estudos de pesquisa, ajudou a produzir ativadores Nrf2 para uso como suplementos dietéticos. O escopo de nossas informações é limitado a questões de quiropraxia e saúde da coluna vertebral. Para discutir o assunto, sinta-se à vontade para perguntar ao Dr. Jimenez ou entre em contato conosco em 915-850-0900 .
Curated pelo Dr. Alex Jimenez
Discussão Adicional do Tópico: Aliviar a Dor no Joelho sem Cirurgia
A dor no joelho é um sintoma bem conhecido que pode ocorrer devido a uma variedade de lesões e / ou condições no joelho, incluindo lesões esportivas. O joelho é uma das articulações mais complexas do corpo humano, pois é formado pela intersecção de quatro ossos, quatro ligamentos, vários tendões, dois meniscos e cartilagem. De acordo com a Academia Americana de Médicos de Família, as causas mais comuns de dor no joelho incluem subluxação patelar, tendinite patelar ou joelho de saltador e doença de Osgood-Schlatter. Embora a dor no joelho seja mais provável de ocorrer em pessoas com mais de 60 anos de idade, a dor no joelho também pode ocorrer em crianças e adolescentes. A dor no joelho pode ser tratada em casa seguindo os métodos do RICE, no entanto, lesões graves no joelho podem exigir atenção médica imediata, incluindo tratamento quiroprático.
Os antioxidantes são cientificamente referidos como compostos que restringem o processo de oxidação no corpo humano, que, se não for controlado, pode criar radicais livres que podem desenvolver numerosas reações em cadeia que podem causar danos celulares. Felizmente, o corpo humano pode criar tais mecanismos imunes embutidos, no entanto, quando a montagem de espécies reativas de oxigênio, ou ROS, não consegue ser neutralizada, visualiza uma pequena chama que fica fora de controle quando infundida com oxigênio, o dano está prestes a ocorrer .
Para continuar a expandir a metáfora da chama, o produto final de não ter a capacidade de neutralizar o impacto das espécies reativas de oxigênio é o dano, bem como a inflamação, em outras palavras, o corpo humano está literalmente em chamas. O fantástico é que existem antioxidantes que podem ajudar tremendamente a combater este problema de saúde e este antioxidante é a glutationa. Embora encontrado em 1889, o efeito antioxidante da glutationa tornou-se um dos tópicos mais interessantes em pesquisas modernas.
Mestre dos Antioxidantes: Glutationa
A substância poderosa é um tripeptídeo que se desenvolve a partir da cisteína, ácido glutâmico e glicina. Devido à sua capacidade de proteger o corpo humano contra a criação de radicais livres, a glutationa pode ajudar a promover um sistema imunológico saudável. Baseado em Relatórios Científicos no 2015, foi determinado que a capacidade da glutationa para funcionar sinergicamente com peroxiredina e catalase ajuda a proteger as células contra o peróxido de hidrogênio. Esta fórmula sinérgica funciona contra espécies reativas de oxigênio, ou ROS. A glutationa, a peroxidredina e a catalase são elementos essenciais no aumento da homeostase celular, que é um processo essencial de células, tecidos e órgãos saudáveis.
Além disso, a glutationa aumenta a estrutura e a função geral do sistema imunológico, utilizando seu importante efeito nas funções linfocitárias. De acordo com Departamento de Imunoquímica, complementando adequadamente os níveis de glutationa no corpo humano pode aumentar consideravelmente as reações imunes. A título de exemplo, dois ensaios randomizados controlados com placebo demonstraram que o tratamento terapêutico de pacientes imunocomprometidos com N-acetilcisteína, ou NAC, resultou, em ambos os casos, em um crescimento substancial na maioria dos processos imunológicos que incluíram um rejuvenescimento completo. de atividade celular natural killer. A N-acetil-cisteína, ou NAC, usa o enxofre da glutationa e a combina com moléculas venenosas, que então se tornam solúveis em água e são liberadas no corpo humano.
A glutationa também tem a capacidade de revitalizar o ácido lipóico, bem como de reciclar a Vitamina C e E, que são necessárias para iniciar certos processos do sistema enviando elétrons para neutralizar os radicais livres. Baseado em um estudo de pesquisa de PLoS ONE, glutationa afetou pacientes com diabetes metillus, ou T2DM e mycobacterium tuberculosis. Normalmente, indivíduos com sistemas imunológicos fracos têm uma tendência a mostrar maior exposição a M. tuberculosis, tuberculose ou micobacterium tuberculosis, doença ou infecção. Além disso, indivíduos com diabetes tipo 2 metillus, ou T2DM, são duas a três vezes mais propensos à TB do que pessoas sem T2DM. O estudo também sugeriu que o aumento dos níveis de glutationa em macrófagos isolados de pacientes com T2DM levou a um melhor controle da doença ou infecção por M.Tb. Estes resultados demonstram que níveis mais baixos de glutationa em pacientes com T2DM contribuem para uma maior chance de doença ou infecção por M. tb. Além disso, depende de Dietro Ghezzi em Brighton e Sussex Medical School, o estresse oxidativo pode, em última análise, causar má estrutura e função do sistema imunológico.
Felizmente, a glutationa desempenha um papel essencial no fortalecimento e controle da imunidade. Por exemplo, a glutationa é essencial para processos inatos e adaptativos dentro do sistema imunológico, incluindo proliferação de linfócitos T, atividade fagocítica de neutrófilos polimorfonucleares e funções de células dendríticas, que podem ser fundamentais porque são constituídos de células apresentadoras de antígenos. . imunidade celular mediados pelo inclui antigios de protea que inicialmente comecem a degenerar nas vesículas endocíticas de macrófagos e células dendríticas, por conseguinte, os péptidos mais pequenos são demonstradas na superfície para activar a proliferação de células T específicas de antigénio. Além disso, a glutationa ajuda na criação de citocinas, e é necessário manter a produção de interferon-gama pelas células dendríticas, o que é importante para a proteção contra patógenos intracelulares, incluindo micobactérias.
N-acetil-cisteína, ou NAC, cientificamente referido como o precursor da glutationa, também é um antioxidante celular muito poderoso usado como um antioxidante eliminador de radicais livres. Comumente reconhecido por seu papel em evitando toxicidade por acetaminofeno, NAC, ou N-acetil-cisteína, demonstrou possuir vários benefícios de saúde e bem-estar. De acordo com Jornal de Células, O NAC ajuda a suportar uma resposta inflamatória saudável e pode impactar positivamente os trabalhos a termo e pré-termo em humanos. O estudo concluiu que em mulheres com parto prematuro anterior e vaginose bacteriana, 0.6 grama de NAC por dia tomado por via oral juntamente com progesterona após a semana 16 de gravidez protegeu contra a recorrência do nascimento prematuro e melhorou o resultado neonatal. Em conclusão, os efeitos positivos do NAC na construção muscular também foram detectados. Após três minutos de contrações persistentes, houve aumento de 15 por cento, demonstrando como o NAC desempenha um papel fundamental na melhoria da construção muscular e na redução da fadiga geral durante o trabalho de parto.
Os pesquisadores também descobriram que o NAC, ou N-acetil-cisteína, pode beneficiar aqueles que têm a síndrome do ovário policístico, ou SOP. A SOP, ou síndrome do ovário policístico, é uma doença comum relacionada às glândulas endócrinas que afeta aproximadamente 5 a 10 por cento das mulheres em idade reprodutiva. Nesses pacientes, há um risco maior de sofrer de síndrome metabólica, em que o uso de NAC ajudou a restaurar os níveis saudáveis de insulina e a sensibilidade.
Insight do Dr. Alex Jimenez
Glutationa tem sido referido como o "mestre dos antioxidantes", devido ao seu papel fundamental na obtenção e manutenção da saúde e bem-estar geral. Enquanto o corpo humano é capaz de produzir sua própria glutationa, má nutrição, poluição, toxinas, uso excessivo de drogas e / ou medicamentos, estresse, trauma, envelhecimento, doenças e radiação podem diminuir nossos níveis naturais de glutationa. Isso pode, por sua vez, tornar os indivíduos mais suscetíveis a danos celulares causados por estresse oxidativo, radicais livres, infecções e câncer. Suplementação de glutationa pode, portanto, ter enormes benefícios para o corpo humano. Juntamente com opções alternativas de tratamento, como a quiropraxia, os níveis de glutationa podem ser novamente regulados para melhorar o bem-estar.
Além disso, os profissionais de saúde sugeriram a implementação do uso da suplementação de glutationa juntamente com outras opções alternativas de tratamento, como cuidados quiropráticos, para melhorar ainda mais a saúde e bem-estar geral. Antioxidantes são importantes para manter o máximo bem-estar, bem como para inibir a reação em cadeia de radicais livres que causam dano ou dano celular. Antioxidantes poderosos como a glutationa, como mencionado acima, ajudam a regular o desenvolvimento desses radicais livres e proporcionam uma resposta mais saudável do sistema imunológico. Estudos de pesquisa descobriram que cuidados quiropráticos também pode desempenhar um papel essencial neste processo, impulsionando naturalmente a atividade de antioxidantes no corpo humano. A quiropraxia é uma abordagem de tratamento seguro e eficaz que utiliza ajustes da coluna vertebral e manipulações manuais para corrigir desalinhamentos vertebrais ou subluxações, a fim de permitir que o corpo humano para curar-se naturalmente sem o uso de drogas / medicamentos e / ou intervenções cirúrgicas.
Finalmente, os antioxidantes demonstram suas propriedades biológicas por meio de uma grande quantidade de benefícios à saúde, que podem ser necessários agora mais do que nunca com o ataque cada vez maior de estresse, doenças e poluição em nosso mundo moderno, que contribuem para danos e / ou danos às células . A glutationa e seu precursor, NAC, ou N-acetil-cisteína, continuam a mostrar seu status poderoso no reino dos antioxidantes. Junto com opções alternativas de tratamento, como a quiropraxia, as pessoas podem tirar proveito de todos os benefícios que este poderoso antioxidante tem a oferecer. O escopo de nossas informações é limitado à quiropraxia, bem como a lesões e condições da coluna vertebral. Para discutir o assunto, sinta-se à vontade para perguntar ao Dr. Jimenez ou entre em contato conosco em 915-850-0900 .
Curated pelo Dr. Alex Jimenez
Tópicos adicionais: Dor nas costas
Dor nas costas é uma das causas mais comuns de incapacidade e dias perdidos no trabalho em todo o mundo. De fato, a dor nas costas tem sido atribuída como a segunda razão mais comum para visitas a consultórios, superada apenas por infecções respiratórias superiores. Aproximadamente 80 por cento da população experimentará algum tipo de dor nas costas pelo menos uma vez ao longo da vida. A coluna é uma estrutura complexa composta de ossos, articulações, ligamentos e músculos, entre outros tecidos moles. Por causa disso, lesões e / ou condições agravadas, como hérnia de discos, pode eventualmente levar a sintomas de dor nas costas. Lesões esportivas ou acidentes automobilísticos geralmente são a causa mais frequente de dor nas costas, no entanto, às vezes, o mais simples dos movimentos pode ter resultados dolorosos. Felizmente, opções alternativas de tratamento, como quiropraxia, podem ajudar a aliviar a dor nas costas através do uso de ajustes espinhais e manipulações manuais, melhorando o alívio da dor.
Chiropractor baseado na ciência Dr. Alexander Jimenez olha para estresse oxidativo, o que é, como isso afeta o corpo e a defesa antioxidante para remediar a situação.
Esra Birben PhD, 1 Umit Murat Sahiner MD, 1 Cansin Sackesen MD, 1 Serpil Erzurum MD, 2 e Omer Kalayci, MD1
Resumo: As espécies reativas de oxigênio (ROS) são produzidas por organismos vivos como resultado do metabolismo celular normal e de fatores ambientais, como poluentes do ar ou fumaça de cigarro. ROS são moléculas altamente reativas e podem danificar estruturas celulares como carboidratos, ácidos nucléicos, lipídios e proteínas e alterar suas funções. A mudança no equilíbrio entre oxidantes e antioxidantes em favor dos oxidantes é denominada “estresse oxidativo”. A regulação do estado de redução e oxidação (redox) é crítica para a viabilidade celular, ativação, proliferação e função do órgão. Os organismos aeróbicos têm sistemas antioxidantes integrados, que incluem antioxidantes enzimáticos e não enzimáticos que geralmente são eficazes no bloqueio dos efeitos prejudiciais das ROS. No entanto, em condições patológicas, os sistemas antioxidantes podem ser sobrecarregados. O estresse oxidativo contribui para muitas condições e doenças patológicas, incluindo câncer, distúrbios neurológicos, aterosclerose, hipertensão, isquemia / perfusão, diabetes, síndrome do desconforto respiratório agudo, fibrose pulmonar idiopática, doença pulmonar obstrutiva crônica e asma. Nesta revisão, resumimos os sistemas oxidante e antioxidante celular e discutimos os efeitos e mecanismos celulares do estresse oxidativo.
Palavras-chave: antioxidante, oxidante, estresse oxidativo, espécies reativas de oxigênio, redox
(WAO Journal 2012; 5: 9 19)
As espécies reativas de oxigênio (ROS) são produzidas por organismos vivos como resultado do metabolismo celular normal. Em concentrações baixas a moderadas, eles funcionam em processos celulares fisiológicos, mas em altas concentrações, eles produzem modificações adversas nos componentes celulares, como lipídios, proteínas e DNA.1 6 A mudança no equilíbrio entre oxidante / antioxidante em favor de oxidantes é denominado estresse oxidativo. O estresse oxidativo contribui para muitas condições patológicas, incluindo câncer, distúrbios neurológicos, 7 10 aterosclerose, hipertensão, isquemia / perfusão, 11 14 diabetes, síndrome do desconforto respiratório agudo, fibrose pulmonar idiopática, doença pulmonar obstrutiva crônica , 15 e asma.16 21 Os organismos aeróbicos têm sistemas antioxidantes integrados, que incluem antioxidantes enzimáticos e não enzimáticos que geralmente são eficazes no bloqueio dos efeitos prejudiciais das ROS. No entanto, em condições patológicas, os sistemas antioxidantes podem ser sobrecarregados. Nesta revisão, resumimos os sistemas oxidantes e antioxidantes celulares e a regulação do estado de redução e oxidação (redox) em estados de saúde e doença.
OXIDANTES
Fontes Endógenas de ROS
Os ROS são produzidos a partir de oxigênio molecular como resultado do metabolismo celular normal. Os ROS podem ser divididos em grupos 2: radicais livres e não-radicais. As moléculas que contêm um ou mais elétrons não emparedados e, portanto, proporcionam reatividade à molécula são chamadas de radicais livres. Quando os radicais livres 2 compartilham seus elétrons não pareados, as formas não-radicais são criadas. Os ROS principais de 3 que são de significância fisiológica são o anião superóxido (O22.), O radical hidroxilo (OH) e o peróxido de hidrogênio (H2O2). Os ROS estão resumidos na Tabela 1.
O ânion superóxido é formado pela adição de 1 elétron ao oxigênio molecular.22 Esse processo é mediado pela nicotina adenina dinucleotídeo fosfato [NAD (P) H] oxidase ou xantina oxidase ou pelo sistema de transporte de elétrons mitocondrial. O principal local para a produção de ânion superóxido é a mitocôndria, a maquinaria da célula para produzir trifosfato de adenosina. Normalmente, os elétrons são transferidos através da cadeia de transporte de elétrons mitocondrial para a redução do oxigênio em água, mas aproximadamente 1 a 3% de todos os elétrons vazam do sistema e produzem superóxido. A NAD (P) H oxidase é encontrada em leucócitos polimorfonucleares, monócitos e macrófagos. Após a fagocitose, essas células produzem uma explosão de superóxido que leva à atividade bactericida. O superóxido é convertido em peróxido de hidrogênio pela ação das superóxido dismutases (SODs, EC 1.15.1.1). O peróxido de hidrogênio se difunde facilmente pela membrana plasmática. O peróxido de hidrogênio também é produzido pela xantina oxidase, aminoácido oxidase e NAD (P) H oxidase 23,24 e em peroxissomos pelo consumo de oxigênio molecular em reações metabólicas. Em uma sucessão de reações chamadas reações de Haber Weiss e Fenton, H2O2 pode se decompor em OH2 na presença de metais de transmissão como Fe21 ou Cu21.25
Fe31 + .O2 ? Fe2 + O2 Haber Weiss
Fe2 + H2O2 ? Fe3 + OH + .OH Reação de Fenton
O próprio O 2 também pode reagir com H2 O2 e gerar OH .26,27 O radical hidroxila é o mais reativo das ROS e pode danificar proteínas, lipídios, carboidratos e DNA. Ele também pode iniciar a peroxidação lipídica ao retirar um elétron dos ácidos graxos poliinsaturados.
As enzimas granulocíticas expandem ainda mais a reatividade de H2O2 via peroxidase de eosinófilos e mieloperoxidase (MPO). Em neutrófilos ativados, H2O2 é consumido por MPO. Na presença de íon cloreto, H2O2 é convertido em ácido hipocloroso (HOCl). O HOCl é altamente oxidativo e desempenha um papel importante na morte dos patógenos nas vias aéreas.28 No entanto, o HOCl também pode reagir com o DNA e induzir interações da proteína DNA e produzir produtos de oxidação da pirimidina e adicionar cloreto às bases do DNA.29,30 Peroxidase de eosinófilos e a MPO também contribuem para o estresse oxidativo por modificação de proteínas por halogenações, nitração e ligações cruzadas de proteínas por meio de radicais tirosil.31 33
Outros radicais livres derivados de oxigênio são os radicais peroxil (ROO $). A forma mais simples destes radicais é o radical hidroperxílico (HOO $) e tem papel na peroxidação de ácidos gordos. Os radicais livres podem desencadear reações em cadeia da peroxidação lipídica, abstraindo um átomo de hidrogênio de um carbono de metileno de cadeia lateral. O radical lipídico então reage com oxigênio para produzir radical peroxilo. O radical peroxilo inicia uma reação em cadeia e transforma os ácidos gordurosos poliinsaturados em hidroperóxidos lipídicos. Os hidroperóxidos lipídicos são muito instáveis e facilmente decompostos em produtos secundários, como aldeídos (como 4-hidroxi-2,3-não-real) e malondialdeídos (MDAs). Os isoprostanos são outro grupo de produtos de peroxidação lipídica que são gerados através da peroxidação do ácido araquidônico e também foram encontrados em níveis elevados de plasma e condensados respiratórios de asmáticos. 34,35 A peroxidação de lipídios perturba a integridade das membranas celulares e leva ao rearranjo da estrutura da membrana .
O peróxido de hidrogênio, o radical superóxido, a glutationa oxidada (GSSG), MDAs, isoprostanos, carbonilos e nitrotirosina podem ser facilmente medidos a partir de amostras de lavagem de plasma, sangue ou broncoalveolar como biomarcadores de oxidação por ensaios padronizados.
Fonte Exógena de Oxidantes
Fumaça de cigarro
A fumaça de cigarro contém muitos oxidantes e radicais livres e compostos orgânicos, como superóxido e óxido nítrico. 36 Além disso, a inalação da fumaça do cigarro no pulmão também ativa alguns mecanismos endógenos, como o acúmulo de neutrófilos e macrófagos, o que aumenta ainda mais a lesão oxidante .
Exposição ao ozônio
A exposição ao ozônio pode causar peroxidação lipídica e induzir influxo de neutrófilos no epitélio das vias aéreas. A exposição a curto prazo ao ozônio também causa a liberação de mediadores inflamatórios, como MPO, proteínas catiônicas de eosinófilos e também lactato desidrogenase e albumina. 37 Mesmo em indivíduos saudáveis, a exposição ao ozônio causa uma redução nas funções pulmonares. 38 Cho et al39 demonstraram que matéria particulada (mistura de partículas sólidas e gotículas líquidas suspensas no ar) catalisa a redução de oxigênio.
Hiperoxia
Hiperoxia refere-se a condições de níveis mais elevados de oxigênio do que a pressão parcial normal de oxigênio nos pulmões ou outros tecidos corporais. Isso leva a uma maior produção de espécies reativas de oxigênio e nitrogênio. 40,41
Radiação ionizante
A radiação ionizante, na presença de O2, converte o radical hidroxila, superóxido e radicais orgânicos em peróxido de hidrogênio e hidroperóxidos orgânicos. Essas espécies de hidroperóxidos reagem com íons metálicos redox ativos, como Fe e Cu, por meio de reações de Fenton e, assim, induzem estresse oxidativo.42,43 Narayanan e cols.44 mostraram que fibroblastos que foram expostos a partículas alfa tiveram aumentos significativos em O2 2 intracelular e H2O2 produção via NADPH oxidase ligada à membrana plasmática.44 Moléculas de transdução de sinal, como quinase 1 e 2 regulada por sinal extracelular (ERK1 / 2), quinase N-terminal c-Jun (JNK) e p38, e fatores de transcrição, como proteína ativadora-1 (AP-1), fator nuclear-kB (NF-kB) e p53, são ativados, o que resulta na expressão de genes relacionados à resposta à radiação.45 50 fótons ultravioleta A (UVA) desencadeiam reações oxidativas por excitação de fotossensibilizadores endógenos, como porfirinas, NADPH oxidase e riboflavinas. 8-Oxo-7,8-dihidroguanina (8-oxoGua) é o principal produto de oxidação do DNA mediado por UVA formado pela oxidação do radical OH, oxidantes de 1 elétron e oxigênio singlete que reage principalmente com a guanina.51 A formação de guanina Foi demonstrado que o cátion radical no DNA isolado ocorre de forma eficiente por meio do efeito direto da radiação ionizante.52,53 Após a exposição à radiação ionizante, o nível intracelular de glutationa (GSH) diminui por um curto prazo, mas depois aumenta novamente.54
Ions de metais pesados
Os íons de metais pesados, como ferro, cobre, cádmio, mercúrio, níquel, chumbo e arsênico, podem induzir a geração de radicais reativos e causar danos celulares através da depleção de atividades enzimáticas através da peroxidação lipídica e reação com proteínas nucleares e DNA.55
Um dos mecanismos mais importantes de geração de radicais livres mediada por metal é por meio de uma reação do tipo Fenton. O íon superóxido e o peróxido de hidrogênio podem interagir com metais de transição, como ferro e cobre, por meio da reação de Haber Weiss / Fenton catalisada por metal para formar radicais OH.
Além dos mecanismos do tipo Fenton e do tipo Haber Weiss, certos íons metálicos podem reagir diretamente com moléculas celulares para gerar radicais livres, como os radicais tiol, ou induzir vias de sinalização celular. Esses radicais também podem reagir com outras moléculas de tiol para gerar O22 .. O22. é convertido em H2O2, o que causa a geração adicional de radicais de oxigênio. Alguns metais, como o arsenito, induzem a formação de ROS indiretamente pela ativação de sistemas produtores de radicais nas células.56
O arsênio é um elemento altamente tóxico que produz uma variedade de ROS, incluindo superóxido (O2 2), oxigênio singlete (1O2), radical peroxil (ROO), óxido nítrico (NO), peróxido de hidrogênio (H2O2) e radicais peroxil dimetilarsínico [( CH3) 2AsOO] .57 59 Os compostos de arsênio (III) podem inibir enzimas antioxidantes, especialmente as enzimas dependentes de GSH, como glutationa-S-transferases (GSTs), glutationa peroxidase (GSH-Px) e GSH redutase, via ligação - em seus grupos sulfidrila ( SH ).60,61
O chumbo aumenta a peroxidação lipídica. 62 As diminuições significativas na atividade de SOD de tecido e GPx cerebral foram relatadas após a exposição ao chumbo.63,64 A substituição de zinco, que serve como cofator para muitas enzimas por chumbo, leva à inativação de tais enzimas. A exposição ao chumbo pode causar inibição da GST ao afetar os teolos de tecidos.
Os ROS gerados por reações catalisadas por metal podem modificar as bases de DNA. Três substituições de base, G / C, G / T e C / T podem ocorrer como resultado do dano oxidativo por íons metálicos, como Fe21, Cu21 e Ni21. Reid et al65 mostraram que G / C foi produzido predominantemente por Fe21 enquanto a substituição de C / T era por Cu21 e Ni21.
ANTIOXIDANTES
O corpo humano está equipado com uma variedade de antioxidantes que servem para contrabalançar o efeito dos oxidantes. Para todos os efeitos práticos, estes podem ser divididos em categorias 2: enzimática (Tabela 2) e não enzimática (Tabela 3).
Antioxidantes enzimáticos
Os principais antioxidantes enzimáticos dos pulmões são SODs (EC 1.15.1.11), catalase (EC 1.11.1.6) e GSH-Px (EC 1.11.1.9). Além dessas enzimas principais, outros antioxidantes, incluindo heme oxigenase-1 (EC 1.14.99.3) e proteínas redox, tais como tioredoxinas (TRXs, EC 1.8.4.10), peroxiredoxinas (PRXs, EC 1.11.1.15) e glutaredoxinas, também foram encontrados para desempenham papéis cruciais nas defesas antioxidantes pulmonares.
Uma vez que o superóxido é o ROS primário produzido a partir de uma variedade de fontes, a sua dismutação pelo SOD é de primordial importância para cada célula. Todas as formas 3 de SOD, isto é, CuZn-SOD, Mn-SOD e EC-SOD, são amplamente expressas no pulmão humano. O Mn-SOD está localizado na matriz mitocondria. O EC-SOD é principalmente localizado na matriz extracelular, especialmente em áreas contendo altas quantidades de fibras de colágeno tipo I e em torno de vasos pulmonares e sistêmicos. Também foi detectado no epitélio brônquico, no epitélio alveolar e nos macrófagos alveolares. 66,67 Em geral, CuZn-SOD e Mn-SOD são geralmente pensados para atuar como eliminadores a granel de radicais superóxido. O nível de CE-SOD relativamente elevado no pulmão com a sua ligação específica aos componentes da matriz extracelular pode representar um componente fundamental da proteção da matriz pulmonar. 68
H2O2 que é produzido pela ação de SODs ou a ação de oxidases, como xantina oxidase, é reduzida a água por catalase e GSH-Px. A catalase existe como um tetramer composto por monómeros 4 idênticos, cada um dos quais contém um grupo heme no local ativo. A degradação de H2O2 é realizada através da conversão entre conformações 2 de catalase-ferricatalase (ferro coordenado com água) e composto I (ferro complexado com um átomo de oxigênio). A catalase também se liga ao NADPH como um equivalente de redução para prevenir a inativação oxidativa da enzima (formação do composto II) por H2O2, pois é reduzida à água. 69
Enzimas no ciclo redox responsável pela redução de H2O2 e hidroperóxidos lipídicos (gerados como resultado da peroxidação lipídica da membrana) incluem o GSH-Pxs.70. Os GSH-Pxs são uma família de enzimas tetraméricas que contêm a única selenocisteína de aminoácidos dentro da sites ativos e usam tióis de baixo peso molecular, como GSH, para reduzir H2O2 e peróxidos lipídicos aos seus álcoois correspondentes. Foram descritos quatro GSH-Pxs, codificados por diferentes genes: GSH-Px-1 (GSH-Px celular) é onipresente e reduz H2O2 e peróxidos de ácidos gordos, mas não lipídios de peroxilo esterificados. 71 Os lípidos esterificados são reduzidos por GSH ligado à membrana -Px-4 (hidroperóxido de fosfolípido GSH-Px), que pode usar vários diferentes tióis de baixo peso molecular como equivalentes de redução. GSH-Px-2 (GSH-Px gastrointestinal) está localizado em células epiteliais gastrointestinais, onde serve para reduzir peróxidos alimentares. 72 GSH-Px-3 (GSH-Px extracelular) é o único membro da família GSH-Px que reside em o compartimento extracelular e acredita-se que seja uma das enzimas antioxidantes extracelulares mais importantes em mamíferos. Destes, o GSH-Px extracelular é mais amplamente investigado no pulmão humano. 73
Além disso, a eliminação de H2O2 está estreitamente associada a várias enzimas contendo tiol, nomeadamente TRXs (TRX1 e TRX2), tioredoxina reductasas (EC 1.8.1.9) (TRRs), PRXs (que são as peroxidases de tiorrecoxina) e glutaredoxins.74
Dois TRXs e TRRs foram caracterizados em células humanas, existentes tanto no citosol como nas mitocôndrias. No pulmão, TRX e TRR são expressos no epitélio e nos macrófagos brônquicos e alveolares. Seis PRX diferentes foram encontrados em células humanas, diferindo em sua compartimentação ultra-estrutural. Estudos experimentais revelaram a importância do PRX VI na proteção do epitélio alveolar. O pulmão humano expressa todos os PRXs no epitélio brônquico, epitélio alveolar e macrófagos. 75 PRX V recentemente foi encontrado para funcionar como uma redutase de peroxinitrito, 76, o que significa que pode funcionar como um composto potencial de proteção no desenvolvimento de lesões pulmonares mediadas por ROS .77
Comum a estes antioxidantes é o requisito de NADPH como um equivalente de redução. NADPH mantém a catalase na forma ativa e é usado como cofator por TRX e GSH redutase (EC 1.6.4.2), que converte GSSG em GSH, um co-substrato para GSH-Pxs. O NADPH intracelular, por sua vez, é gerado pela redução de NADP1 por glicose-6-fosfato desidrogenase, a primeira e enzima limitante de taxa de penetração de fosfato, durante a conversão de glicose-6-fosfato em 6-fosfogluconolactona. Ao gerar NADPH, a glicose-6-fosfato desidrogenase é um determinante crítico da capacidade de tampão GSH citosólica (GSH / GSSG) e, portanto, pode ser considerada uma enzima antioxidante reguladora e essencial. 78,79
GSTs (EC 2.5.1.18), outra família de enzimas antioxidantes, inativa metabólitos secundários, como aldeídos insaturados, epóxidos e hidroperóxidos. Três famílias principais de GSTs foram descritas: GST citosólico, GST mitocondrial, 80,81 e GST microssômico associado à membrana que tem um papel no metabolismo de eicosanóides e GSH.82 Sete classes de GST citosólico são identificadas em mamíferos, designadas Alfa, Mu, Pi, Sigma, Theta, Omega e Zeta.83 86 Durante condições sem estresse, os GSTs de classe Mu e Pi interagem com as cinases Ask1 e JNK, respectivamente, e inibem essas cinases.87 89 Foi demonstrado que GSTP1 se dissocia de JNK em resposta ao estresse oxidativo.89 O GSTP1 também interage fisicamente com PRX VI e leva à recuperação da atividade da enzima PRX por meio da glutationilação da proteína oxidada.90
Antioxidantes não enzimáticos
Os antioxidantes não enzimáticos incluem compostos de baixo peso molecular, como vitaminas (vitaminas C e E), b-caroteno, ácido úrico e GSH, um tripeptídeo (Lg-glutamil-L-cisteinil-L-glicina) que compreende um tiol ( sulfidrilo).
A vitamina C (ácido ascórbico)
A vitamina C solúvel em água (ácido ascórbico) fornece capacidade antioxidante de fase aquosa intracelular e extracelular principalmente por eliminação de radicais livres de oxigênio. Converte os radicais livres da vitamina E de volta à vitamina E. Seus níveis de plasma mostraram diminuir com a idade. 91,92
Vitamina E (a-tocoferol)
A vitamina E lipossolúvel é concentrada no local interior hidrofóbico da membrana celular e é a principal defesa contra a lesão da membrana induzida por oxidantes. A vitamina E doa o elétron ao radical peroxilo, que é produzido durante a peroxidação lipídica. A-Tocoferol é a forma mais ativa de vitamina E e o principal antioxidante ligado à membrana nas células. A vitamina E desencadeia a apoptose das células cancerosas e inibe as formações de radicais livres. 93
Glutationa
GSH é altamente abundante em todos os compartimentos celulares e é o principal antioxidante solúvel. A relação GSH / GSSG é um importante determinante do estresse oxidativo. GSH mostra seus efeitos antioxidantes de várias maneiras. 94 Desintoxica peróxido de hidrogênio e peróxidos lipídicos por meio da ação do GSH-Px. GSH doa seu elétron para H2O2 para reduzi-lo em H2O e O2. GSSG é novamente reduzido em GSH pela redutase GSH que usa NAD (P) H como o doador de elétrons. Os GSH-Pxs também são importantes para a proteção da membrana celular a partir da peroxidação lipídica. A glutationa reduzida doa prótons para lipídios da membrana e os protege de ataques oxidantes. 95
GSH é um cofator para várias enzimas desintoxicantes, como GSH-Px e transferase. Tem um papel na conversão de vitamina C e E de volta às suas formas ativas. O GSH protege as células contra a apoptose ao interagir com as vias de sinalização proapoptótica e antiapoptótica. 94 Também regula e ativa vários fatores de transcrição, como AP-1, NF-kB e Sp-1.
Carotenóides (b-caroteno)
Os carotenóides são pigmentos encontrados nas plantas. Principalmente, o b-caroteno encontrou-se reagir com os radicais de peroxilo (ROO), hidroxilo (OH) e superóxido (O22.). Os carotenóides 96 mostram seus efeitos antioxidantes em baixa pressão parcial de oxigênio, mas podem ter efeitos pró-oxidantes em oxigênio superior Concentrações. 97 Ambos os carotenóides e os ácidos retinóicos (RAs) são capazes de regular os fatores de transcrição. 98 b-Carotene inibe a ativação do NF-kB induzido pelo oxidante e a interleucina (IL) -6 e o fator de necrose tumoral - uma produção. Os carotenóides também afetam a apoptose das células. Os efeitos antiproliferativos da AR foram demonstrados em vários estudos. Este efeito da AR é mediado principalmente por receptores de ácido retinóico e varia entre os tipos celulares. Nas células de carcinoma mamário, o receptor de ácido retinoico mostrou que desencadeia a inibição do crescimento induzindo a prisão do ciclo celular, apoptose ou ambos. 99,100
O EFEITO DO ESTRESSE OXIDATIVO: MECANISMOS GENÉTICOS, FISIOLÓGICOS E BIOQUÍMICOS
O estresse oxidativo ocorre quando o equilíbrio entre antioxidantes e ROS são interrompidos devido à depleção de antioxidantes ou à acumulação de ROS. Quando o estresse oxidativo ocorre, as células tentam neutralizar os efeitos oxidantes e restaurar o equilíbrio redox por ativação ou silenciamento de genes que codificam enzimas defensivas, fatores de transição e proteínas estruturais. 101,102 A relação entre glutationa oxidada e reduzida (2GSH / GSSG) é uma dos determinantes importantes do estresse oxidativo no corpo. A maior produção de ROS no corpo pode alterar a estrutura do DNA, resultar na modificação de proteínas e lipídios, ativação de vários fatores de transcrição induzidos pelo estresse e produção de citocinas pró-inflamatórias e anti-inflamatórias.
Efeitos do estresse oxidativo no DNA
O ROS pode levar a modificações do DNA de várias maneiras, o que envolve degradação de bases, rupturas de ADN de cadeia simples ou dupla, purina, pirimidina ou modificações ligadas ao açúcar, mutações, deleções ou translocações e reticulação com proteínas. A maioria dessas modificações de DNA (Fig. 1) são altamente relevantes para carcinogênese, envelhecimento e doenças neurodegenerativas, cardiovasculares e auto-imunes. A fumaça do tabaco, os metais redox e os metais não redox, como ferro, cádmio, cromo e arsênico, também estão envolvidos na carcinogênese e no envelhecimento, gerando radicais livres ou ligando-se a grupos tiol. A formação de 8-OH-G é o dano de ADN mais conhecido que ocorre por meio do estresse oxidativo e é um biomarcador potencial para a carcinogênese.
As regiões promotoras dos genes contêm sequências de consenso para fatores de transcrição. Esses sítios de ligação do fator de transcrição contêm sequências ricas em GC que são suscetíveis a ataques de oxidantes. A formação de DNA 8-OH-G em locais de ligação de fator de transcrição pode modificar a ligação de fatores de transcrição e, assim, alterar a expressão de genes relacionados, como foi demonstrado para sequências alvo AP-1 e Sp-1 Além de 103-OH-G, 8 -ciclo-8,59-desoxiadenosina (ciclo-dA) também demonstrou inibir a transcrição de um gene repórter em um sistema celular se localizado em uma caixa TATA.29 A proteína de ligação a TATA inicia a transcrição alterando a curvatura do DNA . A ligação da proteína de ligação a TATA pode ser prejudicada pela presença de ciclo-dA.
O estresse oxidativo causa instabilidade de regiões de microssatélites (repetições em tandem curtas). Os íons metálicos ativos Redox, os radicais hidroxílicos aumentam a instabilidade dos microsatérios. 105 Embora as rupturas de DNA de cadeia simples causadas por lesão oxidante possam ser facilmente toleradas pelas células, as rupturas de DNA de cadeia dupla induzidas por radiação ionizante podem ser uma ameaça significativa para a sobrevivência celular. 106
A metilação nas ilhas CpG no DNA é um importante mecanismo epigenético que pode resultar em silenciamento gênico. A oxidação de 5-MeCyt para 5-hidroximetil uracilo (5-OHMeUra) pode ocorrer através de reações de desingação / oxidação de intermediários de timina ou 5-hidroximetil citosina. 107 Além da expressão genética moduladora, a metilação do DNA também parece afetar a organização da cromatina. 108 Os padrões aberrantes de metilação do DNA induzidos por ataques oxidativos também afetam a atividade de reparo do DNA.
Efeitos do estresse oxidativo em lipídios
ROS pode induzir peroxidação lipídica e interromper o arranjo da bicamada lipídica da membrana que pode inativar receptores e enzimas ligados à membrana e aumentar a permeabilidade do tecido.109 Produtos de peroxidação lipídica, como MDA e aldeídos insaturados, são capazes de inativar muitas proteínas celulares por meio da formação de proteínas -ligações.110 112 4-Hidroxi-2-nonenal causa depleção de GSH intracelular e induz a produção de peróxido, 113,114 ativa o receptor do fator de crescimento epidérmico 115 e induz a produção de fibronectina.116 Produtos de peroxidação lipídica, como isoprostanos e substâncias reativas ao ácido tiobarbitúrico , têm sido usados como biomarcadores indiretos de estresse oxidativo, e níveis aumentados foram mostrados no condensado do ar exalado ou no fluido de lavagem broncoalveolar ou no pulmão de pacientes com doença pulmonar obstrutiva crônica ou fumantes.117 119
Efeitos do estresse oxidativo nas proteínas
ROS podem causar fragmentação da cadeia peptídica, alteração da carga elétrica de proteínas, reticulação de proteínas e oxidação de aminoácidos específicos e, portanto, levar ao aumento da suscetibilidade à proteólise por degradação por proteases específicas.120 Resíduos de cisteína e metionina nas proteínas são particularmente mais suscetível à oxidação.121 A oxidação de grupos sulfidrila ou resíduos de metionina de proteínas causa alterações conformacionais, desdobramento de proteínas e degradação.8,121 123 Enzimas que têm metais em ou perto de seus locais ativos são especialmente mais sensíveis à oxidação catalisada por metal. A modificação oxidativa de enzimas demonstrou inibir suas atividades.124,125
Em alguns casos, a oxidação específica das proteínas pode ocorrer. Por exemplo, a metionina pode ser oxidada sulfato de metionina126 e fenilalanina a o-tirosina127; Os grupos sulfidrilo podem ser oxidados para formar ligações dissulfureto; os grupos 128 e carbonilo podem ser introduzidos nas cadeias laterais das proteínas. Os raios gama, oxidação catalisada por metal, HOCl e ozônio podem causar a formação de grupos carbonilo. 129
Efeitos do estresse oxidativo na transdução do sinal
ROS pode induzir a expressão de vários genes envolvidos na transdução de sinal.1,130 Uma alta proporção para GSH / GSSG é importante para a proteção da célula de danos oxidativos. A ruptura dessa proporção causa ativação de fatores de transcrição redox sensíveis, como NF-kB, AP-1, fator nuclear de células T ativadas e fator 1 indutível por hipóxia, que estão envolvidos na resposta inflamatória. A ativação de fatores de transcrição via ROS é obtida por cascatas de transdução de sinal que transmitem a informação de fora para dentro da célula. Receptores de tirosina quinase, a maioria dos receptores de fator de crescimento, como receptor de fator de crescimento epidérmico, receptor de fator de crescimento endotelial vascular e receptor de fator de crescimento derivado de plaquetas, proteína tirosina fosfatases e serina / treonina quinases são alvos de ROS.131 133 Quinases extracelulares reguladas por sinal, JNK e p38, que são os membros da família da proteína quinase ativada por mitogênio e envolvidas em vários processos celulares, incluindo proliferação, diferenciação e apoptose, também podem ser reguladas por oxidantes.
Sob condições de estresse oxidativo, os resíduos de cisteína no sítio de ligação ao DNA de c-Jun, algumas subunidades AP-1 e a quinase kB inibitória sofrem glutatiolação S reversível. Foi relatado que glutaredoxina e TRX desempenham um papel importante na regulação das vias de sinalização sensíveis a redox, como NF-kB e AP-1, proteína quinase ativada por mitogênio p38 e JNK.134 137
NF-kB pode ser ativado em resposta a condições de estresse oxidativo, como ROS, radicais livres e irradiação UV.138 A fosforilação de IkB libera NF-kB e permite que ele entre no núcleo para ativar a transcrição do gene.139 Várias quinases têm foi relatado que fosforila IkBs nos resíduos de serina. Essas quinases são os alvos dos sinais oxidativos para a ativação do NF-kB.140. Os agentes redutores aumentam a ligação do NF-kB ao DNA, enquanto os agentes oxidantes inibem a ligação do NF-kB ao DNA. TRX pode exercer 2 ações opostas na regulação de NF-kB: no citoplasma, ele bloqueia a degradação de IkB e inibe a ativação de NF-kB, mas aumenta a ligação ao DNA de NF-kB no núcleo.141 Ativação de NF-kB por meio de degradação relacionada à oxidação de IkB resulta na ativação de vários genes relacionados à defesa antioxidante. O NF-kB regula a expressão de vários genes que participam da resposta imune, como IL-1b, IL-6, fator de necrose tumoral-a, IL-8 e várias moléculas de adesão.142,143 NF-kB também regula a angiogênese e proliferação e diferenciação de células.
O AP-1 também é regulado pelo estado redox. Na presença de H2O2, alguns íons metálicos podem induzir a ativação de AP-1. O aumento da proporção de GSH / GSSG aumenta a ligação de AP-1 enquanto o GSSG inibe a ligação do DNA da ligação de DNA AP-1.144 do heterodímero Fos / Jun é aumentada pela redução de uma única cisteína conservada no domínio de ligação ao DNA de cada uma das as proteínas, 145, enquanto a ligação de DNA de AP-1 pode ser inibida pelo GSSG em muitos tipos de células, sugerindo que a formação de ligações dissulfureto por resíduos de cisteína inibe a ligação do DNA AP-1. A transdução do sinal 146,147 através do estresse oxidativo é resumida na Figura 2.
CONCLUSÕES
O estresse oxidativo pode surgir da superprodução de ROS por reações metabólicas que usam oxigênio e deslocam o equilíbrio entre oxidante /antioxidante status a favor dos oxidantes. Os ROS são produzidos por atividades metabólicas celulares e fatores ambientais, como poluentes do ar ou fumaça de cigarro. ROS são moléculas altamente reativas devido a elétrons não emparelhados em sua estrutura e reagem com várias macromoléculas biológicas em células, como carboidratos, ácidos nucleicos, lipídios e proteínas, e alteram suas funções. O ROS também afeta a expressão de vários genes por upregulação de fatores de transcrição redox-sensíveis e remodelação da cromatina por alteração na acetilação / desacetilação de histonas. O regulamento do estado redox é crítico para viabilidade celular, ativação, proliferação e função orgânica.
REFERÊNCIAS
1. Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Radicais livres, metais e antioxidantes no câncer induzido por estresse oxidativo. Chem Biol Interact. 2006; 160: 1 40.
2. Halliwell B, Gutteridge JMC. Radicais livres em biologia e medicina. 3rd ed. Nova York: Oxford University Press; 1999.
3. Marnett LJ. Peroxidação lipídica Dano de DNA por malondialdeído. Mutat Res. 1999; 424: 83 95.
4. Siems WG, Grune T, formação de Esterbauer H. 4-Hydroxynonenal durante a isquemia e reperfusão do intestino delgado de rato. Life Sci. 1995; 57: 785 789.
5. Stadtman ER. Papel das espécies oxidantes no envelhecimento. Curr Med Chem. 2004; 11: 1105 1112.
6. Wang MY, Dhingra K, Hittelman WN, Liehr JG, deAndrade M, Li DH. Adutos putativos de malondialdeído DNA induzidos por peroxidação lipídica em tecidos mamários humanos. Cancer Epidemiol Biomarkers Prev. 1996; 5: 705 710.
7. Jenner P. Estresse oxidativo na doença de Parkinson. Ann Neurol. 2003; 53: S26 S36.
8. Lyras L, Cairns NJ, Jenner A, Jenner P, Halliwell B. Uma avaliação do dano oxidativo a proteínas, lipídios e DNA no cérebro de pacientes com doença de Alzheimer. J Neurochem. 1997; 68: 2061 2069.
9. Sayre LM, Smith MA, Perry G. Química e bioquímica do estresse oxidativo em doenças neurodegenerativas. Curr Med Chem. 2001; 8: 721 738.
10. Toshniwal PK, Zarling EJ. Evidência de aumento da peroxidação lipídica na esclerose múltipla. Neurochem Res. 1992; 17: 205 207.
11. Dhalla NS, Temsah RM, Netticadan T. Role of oxidative stress in cardiovascular disease. J Hypertens. 2000; 18: 655 673.
12. Kasparova S, Brezova V, Valko M, Horecky J, Mlynarik V, et al. Estudo do estresse oxidativo em um modelo de hipoperfusão cerebral crônica em ratos. Neurochem Int. 2005; 46: 601 611.
13. Kerr S, Brosnan MJ, McIntyre M, Reid JL, Dominiczak AF, Hamilton CA. A produção de ânion superóxido é aumentada em um modelo de hipertensão genética: papel do endotélio. Hipertensão. 1999; 33: 1353 1358.
14. Kukreja RC, Hess ML. O sistema de radicais livres de oxigênio: das equações, passando pelas interações membrana-proteína, até danos e proteção cardiovasculares. Cardiovasc Res. 1992; 26: 641 655.
15. Asami S, Manabe H, Miyake J, Tsurudome Y, Hirano T, et al. O tabagismo induz um aumento no dano oxidativo ao DNA, 8-hidroxidesoxiguanosina, em um local central do pulmão humano. Carcinogênese. 1997; 18: 1763 1766.
16. Andreadis AA, Hazen SL, Comhair SA, Erzurum SC. Eventos oxidativos e nitrosativos na asma. Free Radic Biol Med. 2003; 35: 213 225.
17. Comhair SA, Ricci KS, Arroliga M, Lara AR, Dweik RA, et al. Correlação da deficiência sistêmica de superóxido dismutase com a obstrução ao fluxo de ar na asma. Am J Respir Crit Care Med. 2005; 172: 306 313.
18. Comhair SA, Xu W., Ghosh S, Thunnissen FB, Almasan A, et al. Inativação da superóxido dismutase na fisiopatologia da remodelação e reatividade das vias aéreas asmáticos. Am J Pathol. 2005; 166: 663 674.
19. Dut R, Dizdar EA, Birben E, Sackesen C, Soyer OU, Besler T, Kalayci O. Estresse oxidativo e seus determinantes nas vias aéreas de crianças com asma. Alergia. 2008; 63: 1605 1609.
20. Ercan H, Birben E, Dizdar EA, Keskin O, Karaaslan C, et al. Estresse oxidativo e determinantes genéticos e epidemiológicos da lesão oxidativa na asma infantil. J Allergy Clin Immunol. 2006; 118: 1097 1104.
21. Fitzpatrick AM, Teague WG, Holguin F, Yeh M, Brown LA. Programa de pesquisa em asma severa. A homeostase da glutationa das vias aéreas está alterada em crianças com asma grave: evidência de estresse oxidativo. J Allergy Clin Immunol. 2009; 123: 146 152.
22. Miller DM, Buettner GR, Aust SD. Metais de transição como catalisadores de reações de “autoxidação”. Free Radic Biol Med. 1990; 8: 95 108.
23. Dupuy C, Virion A, Ohayon R, Kaniewski J, D me D, Pommier J. Mecanismo de formação de peróxido de hidrogênio catalisado por NADPH oxidase na membrana plasmática da tireóide. J Biol Chem. 1991; 266: 3739 3743.
24. Granger DN. Papel da xantina oxidase e granulócitos na lesão por isquemia-reperfusão. Am J Physiol. 1988; 255: H1269 H1275.
25. Fenton HJH. Oxidação do ácido tartárico na presença de ferro. J Chem Soc. 1984; 65: 899 910.
26. Haber F, Weiss JJ. A decomposição catalítica do peróxido de hidrogênio por sais de ferro. Proc R Soc Lond Ser A. 1934; 147: 332 351.
27. Liochev SI, Fridovich I. O Haber Weiss pedalou 70 anos depois: uma visão alternativa. Redox Rep. 2002; 7: 55 57.
28. Klebanoff SJ. Mieloperoxidase: amigo e inimigo. J Leukoc Biol. 2005; 77: 598 625.
29. Whiteman M, Jenner A, Halliwell B. Hypochlorous acid-induzida modificações de base em DNA isolado de timo de bezerro. Chem Res Toxicol. 1997; 10: 1240 1246.
30. Kulcharyk PA, Heinecke JW. O ácido hipocloroso produzido pelo sistema mieloperoxidase dos fagócitos humanos induz ligações cruzadas covalentes entre o DNA e a proteína. Bioquímica. 2001; 40: 3648 3656.
31. Brennan ML, Wu W, Fu X, Shen Z, Song W, et al. Uma história de duas controvérsias: definir o papel das peroxidases na formação de nitrotirosina in vivo usando peroxidase de eosinófilos e camundongos deficientes em mieloperoxidase e a natureza das espécies reativas de nitrogênio geradas por peroxidase. J Biol Chem. 2002; 277: 17415 17427.
32. Denzler KL, Borchers MT, Crosby JR, Cieslewicz G, Hines EM, et al. A desgranulação extensiva de eosinófilos e a oxidação das proteínas das vias aéreas mediada pela peroxidase não ocorrem em um modelo de inflamação pulmonar com ovalbumina de camundongo. J Immunol. 2001; 167: 1672 1682.
33. van Dalen CJ, Winterbourn CC, Senthilmohan R, Kettle AJ. Nitrito como substrato e inibidor da mieloperoxidase. Implicações para nitração e produção de ácido hipocloroso em locais de inflamação. J Biol Chem. 2000; 275: 11638 11644.
34. Wood LG, Fitzgerald DA, Gibson PG, Cooper DM, Garg ML. A peroxidação lipídica determinada pelos isoprostanos plasmáticos está relacionada à gravidade da doença na asma leve. Lipids. 2000; 35: 967 974.
35. Montuschi P, Corradi M, Ciabattoni G, Nightingale J, Kharitonov SA, Barnes PJ. Aumento de 8-isoprostano, um marcador de estresse oxidativo, no condensado exalado de pacientes asmáticos. Am J Respir Crit Care Med. 1999; 160: 216 220.
36. Church DF, Pryor WA. Química dos radicais livres da fumaça do cigarro e suas implicações toxicológicas. Perspectiva de saúde da Environ. 1985; 64: 111 126.
37. Hiltermann JT, Lapperre TS, van Bree L, Steerenberg PA, Brahim JJ, et al. Inflamação induzida por ozônio avaliada em escarro e fluido de lavagem brônquica de asmáticos: uma nova ferramenta não invasiva em estudos epidemiológicos sobre poluição do ar e asma. Free Radic Biol Med. 1999; 27: 1448 1454.
38. Nightingale JA, Rogers DF, Barnes PJ. Efeito do ozônio inalado no óxido nítrico exalado, função pulmonar e expectoração induzida em indivíduos normais e asmáticos. Tórax. 1999; 54: 1061 1069.
39. Cho AK, Sioutas C, Miguel AH, Kumagai Y, Schmitz DA, et al. Atividade redox de partículas transportadas pelo ar em diferentes locais na Bacia de Los Angeles. Environ Res. 2005; 99: 40 47.
40. Comhair SA, Thomassen MJ, Erzurum SC. Indução diferencial de glutationa peroxidase extracelular e óxido nítrico sintase 2 em vias aéreas de indivíduos saudáveis expostos a 100% O (2) ou fumaça de cigarro. Am J Respir Cell Mol Biol. 2000; 23: 350 354.
41. Matthay MA, Geiser T, Matalon S, Ischiropoulos H. Oxidant-mediated lung lesão na síndrome de angústia respiratória aguda. Crit Care Med. 1999; 27: 2028 2030.
42. Biaglow JE, Mitchell JB, Held K. A importância do peróxido e do superóxido na resposta de raios-X. Int J Radiat Oncol Biol Phys. 1992; 22: 665 669.
43. Chiu SM, Xue LY, Friedman LR, Oleinick NL. Sensibilização mediada por íons de cobre de locais de fixação da matriz nuclear à radiação ionizante. Bioquímica. 1993; 32: 6214 6219.
44. Narayanan PK, Goodwin EH, Lehnert BE. Partículas alfa iniciam a produção biológica de ânions superóxidos e peróxido de hidrogênio em células humanas. Cancer Res. 1997; 57: 3963 3971.
45. Tuttle SW, Varnes ME, Mitchell JB, Biaglow JE. Sensibilidade a oxidantes químicos e radiação em linhagens celulares CHO deficientes na atividade do ciclo das pentoses oxidativas. Int J Radiat Oncol Biol Phys. 1992; 22: 671 675.
46. Guo G, Yan-Sanders Y, Lyn-Cook BD, Wang T, Tamae D, et al. Manganês
expressão de genes mediada por superóxido dismutase em radiação induzida
respostas adaptativas. Mol Cell Biol. 2003; 23: 2362 2378.
47. Azzam EI, de Toledo SM, Spitz DR, Little JB. Metabolismo oxidativo
modula a transdução de sinal e a formação de micronúcleos no espectador
células de fibroblastos humanos normais irradiados com uma partícula. Cancer Res.
2002; 62: 5436 5442.
48. Leach JK, Van Tuyle G, Lin PS, Schmidt-Ullrich R, Mikkelsen RB.
Geração indutiva de radiação ionizante, dependente das mitocôndrias, de geração reativa
oxigênio / nitrogênio. Cancer Res. 2001; 61: 3894 3901.
49. Dent P, Yacoub A, Fisher PB, Hagan MP, Grant S. MAPK caminhos em
respostas de radiação. Oncogene. 2003; 22: 5885 5896.
50. Wei SJ, Botero A, Hirota K, Bradbury CM, Markovina S, et al. Thioredoxin
a translocação nuclear e a interação com o fator redox-1 ativam o fator de transcrição AP-1 em resposta à radiação ionizante. Cancer Res. 2000; 60: 6688 6695.
51. Cadete J, Douki T, Gasparutto D, Ravanat JL. Dano oxidativo ao DNA: formação, medição e características bioquímicas. Mutat Res. 2003; 531: 5 23.
52. Yokoya A, Cunniffe SM, O Neill P. Efeito da hidratação na indução de quebras de fita e lesões de base em filmes de DNA de plasmídeo por radiação gama. J Am Chem Soc. 2002; 124: 8859 8866.
53. Janssen YM, Van Houten B, Borm PJ, Mossman BT. Respostas de células e tecidos ao dano oxidativo. Lab Invest. 1993; 69: 261 274.
54. Iwanaga M, Mori K, Iida T, Urata Y, Matsuo T, et al. Indução dependente do fator nuclear kappa B da gama glutamilcisteína sintetase por radiação ionizante em células de glioblastoma humano T98G. Free Radic Biol Med. 1998; 24: 1256 1268.
55. Stohs SJ, Bagchi D. Mecanismos oxidativos na toxicidade de íons metálicos. Free Radic Biol Med. 1995; 18: 321 336.
56. Leonard SS, Harris GK, Shi X. Estresse oxidativo induzido por metal e transdução de sinal. Free Radic Biol Med. 2004; 37: 1921 1942.
57. Shi H, Shi X, Liu KJ. Mecanismo oxidativo de toxicidade por arsênio e carcinogênese. Mol Cell Biochem. 2004; 255: 67 78.
58. Pi J, Horiguchi S, Sun Y, Nikaido M, Shimojo N, Hayashi T. Um mecanismo potencial para o comprometimento da formação de óxido nítrico causado pela exposição oral prolongada ao arsenato em coelhos. Free Radic Biol Med.2003; 35: 102 ± 113.
59. Rin K, Kawaguchi K, Yamanaka K, Tezuka M, Oku N, Okada S. As quebras de cadeia de DNA induzidas por ácido dimetilarsínico, um metabólito de arsênico inorgânico, são fortemente aumentadas por radicais de ânions superóxido. Biol Pharm Bull. 1995; 18: 45 58.
60. Waalkes MP, Liu J, Ward JM, Diwan LA. Mecanismos subjacentes à carcinogênese do arsênio: hipersensibilidade de camundongos expostos ao arsênio inorgânico durante a gestação. Toxicologia. 2004; 198: 31 38.
61. Schiller CM, Fowler BA, Woods JS. Efeitos do arsênio na ativação da piruvato desidrogenase. Perspectiva de saúde da Environ. 1977; 19: 205 207.
62. Monterio HP, Bechara EJH, Abdalla DSP. Envolvimento dos radicais livres nas porfirias neurológicas e envenenamento por chumbo. Mol Cell Biochem. 1991; 103: 73 83.
63. Tripathi RM, Raghunath R, Mahapatra S. Sangue chumbo e seu efeito nos níveis de Cd, Cu, Zn, Fe e hemoglobina das crianças. Sci Total Environ. 2001; 277: 161 168.
64. Nehru B, Dua R. O efeito do selênio na dieta sobre a neurotoxicidade do chumbo. J Environ Pathol Toxicol Oncol. 1997; 16: 47 50.
65. Reid TM, Feig DI, Loeb LA. Mutagênese por radicais de oxigênio induzidos por metal. Perspectiva de saúde da Environ. 1994; 102 (supl 3): 57 61.
66. Kinnula VL, Crapo JD. Superóxido dismutases no pulmão e doenças pulmonares humanas. Am J Respir Crit Care Med. 2003; 167: 1600 1619.
67. Kinnula VL. Produção e degradação de metabólitos de oxigênio durante estados inflamatórios no pulmão humano. Curr Drug Targets Inflamm Allergy. 2005; 4: 465 470.
68. Zelko IN, Mariani TJ, Folz RJ. Família multigênica de superóxido dismutase: uma comparação das estruturas, evolução e expressão dos genes CuZn-SOD (SOD1), Mn-SOD (SOD2) e EC-SOD (SOD3). Free Radic Biol Med. 2002; 33: 337 349.
69. Kirkman HN, Rolfo M., Ferraris AM, Gaetani GF. Mecanismos de proteção da catalase por NADPH. Cinética e estequiometria. J Biol Chem. 1999; 274: 13908 13914.
70. Floh L. Glutationa peroxidase. Vida Básica Sci. 1988; 49: 663 668.
71. Arthur JR. As glutationa peroxidases. Cell Mol Life Sei. 2000; 57: 1825 1835.
72. Chu FF, Doroshow JH, Esworthy RS. Expressão, caracterização e distribuição nos tecidos de uma nova glutationa peroxidase dependente de selênio celular, GSHPx-GI. J Biol Chem. 1993; 268: 2571 2576.
73. Comhair SA, Bhathena PR, Farver C, Thunnissen FB, Erzurum SC. Indução extracelular de glutationa peroxidase em pulmões asmáticos: evidências para a regulação redox da expressão em células epiteliais das vias aéreas humanas. FASEB J. 2001; 15: 70 78.
74. Gromer S, Urig S, Becker K. O sistema de tiorredoxina da ciência à clínica. Med Res Rev. 2004; 24: 40 89.
75. Kinnula VL, Lehtonen S, Kaarteenaho-Wiik R., Lakari E, P kk P, et al. Expressão celular específica de peroxiredoxinas no pulmão humano e sarcoidose pulmonar. Tórax. 2002; 57: 157 164.
76. Dubuisson M, Vander Stricht D, Clippe A, Etienne F, Nauser T, et al. A peroxirredoxina 5 humana é uma peroxinitrito redutase. FEBS Lett. 2004; 571: 161 165.
77. Holmgren A. Antioxidant function of thioredoxin and glutaredoxin systems. Sinal antioxidante Redox. 2000; 2: 811 820.
78. Dickinson DA, Forman HJ. Glutationa em defesa e sinalização: lições de um pequeno tiol. Ann NY Acad Sei. 2002; 973: 488 504.
79. Sies H. Glutationa e seu papel nas funções celulares. Free Radic Biol Med. 1999; 27: 916 921.
80. Ladner JE, Parsons JF, Rife CL, Gilliland GL, Armstrong RN. Vias evolutivas paralelas para glutationa transferases: estrutura e mecanismo da enzima kappa da classe mitocondrial rGSTK1-1. Bioquímica. 2004; 43: 52 61.
81. Robinson A, Huttley GA, Booth HS, Board PG. Os estudos de modelagem e bioinformática da glutationa transferase da classe kappa humana prevêem uma nova família da terceira transferase com homologia às isomerases procarióticas de 2-hidroxicromene-2-carboxilato. Biochem J. 2004; 379: 541 552.
82. Jakobsson PJ, Morgenstern R, Mancini J, Ford-Hutchinson A, Persson B. Características estruturais comuns de MAPEGda superfamília generalizada de proteínas associadas à membrana com funções altamente divergentes no metabolismo de eicosanóides e glutationa. Protein Sci. 1999; 8: 689 692.
83. Hayes JD, Pulford DJ. A família do supergene da glutationa S-transferase: regulação da GST e a contribuição das isoenzimas para a quimioproteção do câncer e resistência aos medicamentos. Crit Rev Biochem Mol Biol. 1995; 30: 445 600.
84. Armstrong RN. Estrutura, mecanismo catalítico e evolução das glutationa transferases. Chem Res Toxicol. 1997; 10: 2 18.
85. Hayes JD, McLellan LI. A glutationa e as enzimas dependentes da glutationa representam uma defesa coordenada contra o estresse oxidativo. Free Radic Res. 1999; 31: 273 300.
86. Sheehan D, Meade G, Foley VM, Dowd CA. Estrutura, função e evolução das glutationa transferases: implicações para a classificação de membros não mamíferos de uma antiga superfamília de enzimas. Biochem J. 2001; 360: 1 16.
87. Cho SG, Lee YH, Park HS, Ryoo K., Kang KW, et al. A glutationa S-transferase Mu modula os sinais ativados pelo estresse suprimindo a cinase 1 reguladora do sinal de apoptose. J Biol Chem. 2001; 276: 12749 12755.
88. Dorion S, Lambert H, Landry J. A ativação da via de sinalização de p38 por choque térmico envolve a dissociação de glutationa S-transferase Mu de Ask1. J Biol Chem. 2002; 277: 30792 30797.
89. Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L, et al. Regulação da sinalização JNK por GSTp. EMBO J. 1999; 18: 1321 1334.
90. Manevich Y, Feinstein SI, Fisher AB. A ativação da enzima antioxidante 1-CYS peroxirredoxina requer glutationilação mediada por heterodimerização com pGST. Proc Natl Acad Sci US A. 2004; 101: 3780 3785.
91. Bunker VW. Radicais livres, antioxidantes e envelhecimento. Med Lab Sci. 1992; 49: 299 312.
92. Mezzetti A, Lapenna D, Romano F, Costantini F, Pierdomenico SD, et al. Estresse oxidativo sistêmico e sua relação com a idade e doenças. J Am Geriatr Soc. 1996; 44: 823 827.
93. White E, Shannon JS, Patterson RE. Relação entre vitaminas e
uso de suplemento de cálcio e câncer de cólon. Cancer Epidemiol Biomarkers Prev. 1997; 6: 769 774.
94. Masella R, Di Benedetto R, Vari R, Filesi C, Giovannini C. Novos mecanismos de compostos antioxidantes naturais em sistemas biológicos: envolvimento de glutationa e enzimas relacionadas com a glutationa. J Nutr Biochem. 2005; 16: 577 586.
95. Curello S, Ceconi C, Bigoli C, Ferrari R, Albertini A, Guarnieri C. Alterações no status da glutationa cardíaca após isquemia e reperfusão. Experientia. 1985; 41: 42 43.
96. El-Agamey A, Lowe GM, McGarvey DJ, Mortensen A, Phillip DM, Truscott TG. Química do radical carotenóide e propriedades antioxidantes / pró-oxidantes. Arch Biochem Biophys. 2004; 430: 37 48.
97. Rice-Evans CA, Sampson J, Bramley PM, Holloway DE. Por que esperamos que os carotenóides sejam antioxidantes in vivo? Free Radic Res. 1997; 26: 381 398.
98. Niles RM. Vias de sinalização na quimioprevenção dos retinóides e no tratamento do câncer. Mutat Res. 2004; 555: 81 96.
99. Donato LJ, Noy N. Supressão do crescimento do carcinoma mamário por ácido retinóico: os genes pró-apoptóticos são alvos para o receptor do ácido retinóico e a sinalização da proteína II de ligação ao ácido retinóico celular. Cancer Res. 2005; 65: 8193 8199.
100. Niizuma H, Nakamura Y, Ozaki T, Nakanishi H, Ohira M, et al. O Bcl-2 é um regulador chave para a morte celular apoptótica induzida pelo ácido retinóico no neuroblastoma. Oncogene. 2006; 25: 5046 5055.
101. Dalton TP, Shertzer HG, Puga A. Regulação da expressão do gene por oxigênio reativo. Ann Rev Pharmacol Toxicol. 1999; 39: 67 101.
102. Scandalios JG. Respostas genômicas ao estresse oxidativo. In: Meyers RA, ed. Enciclopédia de Biologia Celular e Molecular e Medicina Molecular. Vol 5. 2ª ed. Weinheim, Alemanha: Wiley-VCH; 2004: 489 512.
103. Ghosh R, Mitchell DL. Efeito do dano oxidativo ao DNA em elementos promotores na ligação do fator de transcrição. Nucleic Acids Res. 1999; 27: 3213 3218.
104. Marietta C, Gulam H, Brooks PJ. Uma única lesão de 8, 50-ciclo-20-desoxiadenosina em uma caixa TATA impede a ligação da proteína de ligação a TATA e reduz fortemente a transcrição in vivo. Reparo de DNA (Amst). 2002; 1: 967 975.
105. Jackson AL, Chen R, Loeb LA. Indução da instabilidade dos microsatérios
por dano oxidativo ao DNA. Proc Natl Acad Sci US A. 1998; 95: 12468 12473.
106. Caldecott KW. Interações proteína-proteína durante o reparo de quebra de fita simples de DNA de mamíferos. Biochem Soc Trans. 2003; 31: 247 251.
107. Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: engines, mutation, and disease. FASEB J. 2003; 17: 1195 1214.
108. Jones PL, Wolffe AP. Relações entre a organização da cromatina e a metilação do DNA na determinação da expressão gênica. Semin Cancer Biol. 1999; 9: 339 347.
109. Girotti AW. Mecanismos de peroxidação lipídica. J Free Radic Biol Med. 1985; 1: 87 95.
110. Siu GM, Draper HH. Metabolismo do malonaldeído in vivo e in vitro. Lipids. 1982; 17: 349 355.
111. Esterbauer H, Koller E, Slee RG, Koster JF. Possível envolvimento do produto da peroxidação lipídica 4-hidroxinonenal na formação de cromolipídeos fluorescentes. Biochem J. 1986; 239: 405 409.
112. Hagihara M, Nishigaki I, Maseki M, Yagi K. Alterações dependentes da idade nos níveis de peróxido de lipídio nas frações de lipoproteína do soro humano. J Gerontol. 1984; 39: 269 272.
113. Keller JN, Mark RJ, Bruce AJ, Blanc E, Rothstein JD, et al. 4- O hidroxinonenal, um produto aldeídico da peroxidação lipídica da membrana, prejudica o transporte de glutamato e a função mitocondrial nos sinaptossomas. Neurociências. 1997; 806: 85 96.
114. Uchida K, Shiraishi M, Naito Y, Torii Y, Nakamura Y, Osawa T. Activation of stress signaling pathways by the end product of lipid peroxidation. O 4-hidroxi-2-nonenal é um potencial indutor da produção de peróxido intracelular. J Biol Chem. 1999; 274: 2234 2242.
115. Suc I, Meilhac O, Lajoie-Mazenc I, Vandaele J, Jurgens G, Salvayre R, Negre-Salvayre A. Activation of EGF receptor by oxidized LDL. FASEB J. 1998; 12: 665 671.
116. Tsukagoshi H, Kawata T, Shimizu Y, Ishizuka T, Dobashi K, Mori M. 4-Hydroxy-2-nonenal aumenta a produção de fibronectina por fibroblastos de pulmão humano IMR-90 parcialmente via ativação do sinal extracelular ligado ao receptor do fator de crescimento epidérmico via da quinase p44 / 42 regulada. Toxicol Appl Pharmacol. 2002; 184: 127 135.
117. Montuschi P, Collins JV, Ciabattoni G, Lazzeri N, Corradi M, Kharitonov SA, Barnes PJ. 8-isoprostano exalado como biomarcador in vivo de estresse oxidativo pulmonar em pacientes com DPOC e fumantes saudáveis. Am J Respir Crit Care Med. 2000; 162: 1175 1177.
118. Morrison D, Rahman I, Lannan S, MacNee W. Permeabilidade epitelial, inflamação e estresse oxidante nos espaços aéreos de fumantes. Am J Respir Crit Care Med. 1999; 159: 473 479.
119. Nowak D, Kasielski M, Antczak A, Pietras T, Bialasiewicz P. Aumento do conteúdo de substâncias reativas ao ácido tiobarbitúrico e peróxido de hidrogênio no condensado da respiração expirada de pacientes com doença pulmonar obstrutiva crônica estável: nenhum efeito significativo do tabagismo. Respir Med. 1999; 93: 389 396.
120. Kelly FJ, Mudway IS. Oxidação de proteínas na interface ar-pulmão. Aminoácidos. 2003; 25: 375 396.
121. Dean RT, Roberts CR, Jessup W. Fragmentation of extracellular and intracellular polypeptides by free radicais. Prog Clin Biol Res. 1985; 180: 341 350.
122. Keck RG. O uso de hidroperóxido de t-butila como uma sonda para a oxidação de metionina em proteínas. Anal Biochem. 1996; 236: 56 62.
123. Davies KJ. Dano e degradação de proteínas por radicais de oxigênio. I. Aspectos gerais. J Biol Chem. 1987; 262: 9895 9901.
124. Stadtman ER. Oxidação de proteínas catalisadas por íons metálicos: mecanismo bioquímico e conseqüências biológicas. Radic Biol Med gratuito.
1990; 9: 315 325.
125. Fucci L, Oliver CN, Coon MJ, Stadtman ER. Inativação de enzimas metabólicas chave por reações de oxidação de função mista: possível implicação no turnover de proteínas e envelhecimento. Proc Natl Acad Sci US A. 1983; 80: 1521-1525.
126. Stadtman ER, Moskovitz J, Levine RL. Oxidação de resíduos de metionina de proteínas: consequências biológicas. Sinal antioxidante Redox. 2003; 5: 577 582.
127. Stadtman ER, Levine RL. Oxidação mediada por radicais livres de aminoácidos livres e resíduos de aminoácidos em proteínas. Aminoácidos. 2003; 25: 207 218.
128. Stadtman ER. Oxidação de proteínas no envelhecimento e doenças relacionadas com a idade. Ann NY Acad Sei. 2001; 928: 22 38.
129. Shacter E. Quantificação e significado da oxidação de proteínas em amostras biológicas. Drug Metab Rev. 2000; 32: 307 326.
130. Poli G, Leonarduzzi G, Biasi F, Chiarpotto E. Oxidative stress and cell signaling. Curr Med Chem. 2004; 11: 1163 1182.
131. Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999; 13: 9 22.
132. Sundaresan M, Yu ZX, Ferrans VJ, Sulciner DJ, Gutkind JS, et al. Regulação da geração de espécies reativas de oxigênio em fibroblastos por Rac1. Biochem J. 1996; 318: 379 382.
133. Sun T, Oberley LW. Regulação redox de ativadores transcricionais. Free Radic Biol Med. 1996; 21: 335 348.
134. Klatt P, Molina EP, De Lacoba MG, Padilla CA, Martinez-Galesteo E, Barcena JA, Lamas S. Redox regulação de c-Jun DNA binding by reversible S-glutathiolation. FASEB J. 1999; 13: 1481 1490.
135. Reynaert NL, Ckless K, Guala AS, Wouters EF, Van der Vliet A, Janssen Heininger
YM. Detecção in situ de proteínas S-glutationiladas após derivatização de cisteína catalisada por glutaredoxina-1. Biochim Biophys Acta. 2006; 1760: 380 387.
136. Reynaert NL, Wouters EF, Janssen-Heininger YM. Modulação de glutaredoxina-1
expressão em um modelo de camundongo de doença alérgica das vias aéreas. Am J Respir Cell Mol Biol. 2007; 36: 147 151.
137. Filomeni G, Rotilio G, Ciriolo MR. Sinalização celular e o sistema redox da glutationa. Biochem Pharmacol. 2002; 64: 1057 1064.
138. Pande V, Ramos MJ. Reconhecimento molecular da prostaglandina J (15) 12,14-desoxidelta (2) pelo fator nuclear kappa B e outras proteínas celulares. Bioorg Med Chem Lett. 2005; 15: 4057 4063.
139. Perkins ND. Integração de vias de sinalização celular com função NF-kappaB e IKK. Nat Rev Mol Cell Biol. 2007; 8: 49 62.
140. Gilmore TD. Introdução ao NF-kappaB: jogadores, percursos, perspectivas. Oncogene. 2006; 25: 6680 6684.
141. Hirota K, Murata M, Sachi Y, Nakamura H, Takeuchi J, Mori K, Yodoi J. Papéis distintos da tioredoxina no citoplasma e no núcleo. Um mecanismo de duas etapas de regulação redox do fator de transcrição NF-kappaB. J Biol Chem. 1999; 274: 27891 27897.
142. Ward PA. Papel do complemento, quimiocinas e citocinas regulatórias na lesão pulmonar aguda. Ann NY Acad Sei. 1996; 796: 104 112.
143. Akira S, Kishimoto A. NF-IL6 e NF-kB na regulação do gene de citocina. Adv Immunol. 1997; 65: 1 46.
144. Meyer M, Schreck R, Baeuerle PA. H2O2 e antioxidantes têm efeitos opostos na ativação de NF-kappa B e AP-1 em células intactas: AP-1 como fator responsivo a antioxidante secundário. EMBO J. 1993; 12: 2005 2015.
145. Abate C, Patel L, Rausher FJ, Curran T. Redox regulação de fos e jun DNA-binding activity in vitro. Ciência. 1990; 249: 1157 1161.
146. Galter D, Mihm S, Droge W. Distinct effects of glutationa disulfide on the nuclear transcription fatores kB and the activator protein-1. Eur J Biochem. 1994; 221: 639 648.
147. Hirota K, Matsui M, Iwata S, Nishiyama A, Mori K, Yodoi J. A atividade transcricional de AP-1 é regulada por uma associação direta entre tioredoxina e Ref-1. Proc Natl Acad Sci US A. 1997; 94: 3633 3638.
A ferramenta Find A Practitioner do IFM é a maior rede de referência em Medicina Funcional, criada para ajudar os pacientes a localizar profissionais de Medicina Funcional em qualquer lugar do mundo. Os Praticantes Certificados IFM são listados em primeiro lugar nos resultados da pesquisa, devido à sua extensa educação em Medicina Funcional