
Neuroinflamação:
Sumário
Várias linhas de evidência apoiam o papel patogênico da neuroinflamação em doenças psiquiátricas. Enquanto as doenças autoimunes sistêmicas são causas bem documentadas de transtornos neuropsiquiátricos, as encefalites auto-imunes sinápticas com sintomas psicóticos geralmente são menos reconhecidas. Paralelamente à ligação entre os sintomas psiquiátricos e a auto-imunidade em doenças auto-imunes, anormalidades neuroimunológicas ocorrem em distúrbios psiquiátricos clássicos (por exemplo, depressão maior, bipolar, esquizofrenia e transtornos obsessivo-compulsivos). As investigações sobre a fisiopatologia destas condições tradicionalmente sublinharam a desregulação dos sistemas glutamatérgicos e monoaminérgicos, mas os mecanismos que causam essas anormalidades neurotransmissoras permanecem evasivos. Revisamos o link entre auto-imunidade e distúrbios neuropsiquiátricos, e as evidências humanas e experimentais que apoiam o papel patogênico da neuroinflamação em distúrbios psiquiátricos clássicos selecionados. Compreender como os sistemas psicossocial, genético, imunológico e neurotransmissor interagem podem revelar pistas patogênicas e ajudar a identificar novas terapias preventivas e sintomáticas.
Palavras-chave:
- Neuroinflamação,
- Psychoneuroimmunology,
- Astrocyte,
- Microglia,
- Cytokines,
- Estresse oxidativo,
- Depressão,
- Transtorno obsessivo-compulsivo
- Transtorno bipolar, esquizofrenia
Conteúdo
Introdução
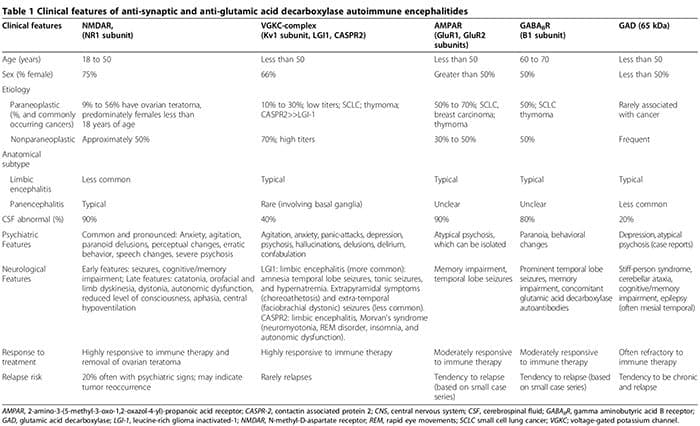
À medida que as anormalidades biológicas são cada vez mais identificadas entre pacientes com transtornos psiquiátricos, a distinção entre doenças neurológicas e psiquiátricas desaparece. Além das doenças sistêmicas auto-imunes associadas a manifestações psiquiátricas (por exemplo, lúpus) [1], mais recentemente, pacientes com psicose isolada aguda foram identificados com encefalite sinimática autoimune (Tabela 1) [2-6]. Esses pacientes freqüentemente são erroneamente diagnosticados com distúrbios psicóticos primários refratários, atrasando o início da imunoterapia efetiva (Tabela 1). Além disso, evidências crescentes apoiam o papel patogênico dos anticorpos anti-neuronais em transtornos neuropsiquiátricos [7].
Separação de distúrbios neurológicos e psiquiátricos, apoiada pela concepção de Descartes da mente como uma entidade ontologicamente distinta e pela reprodutibilidade de anormalidades neuropatológicas, dominou a medicina em 19 e 20 séculos iniciais [8]. Desde então, uma coleção em expansão de causas biológicas reprodutíveis, de neurosífilis, traumatismo craniano, acidente vascular cerebral, tumor, desmielinização e muitos outros causaram complexos de sintomas que se sobrepunham com distúrbios psiquiátricos clássicos [9-11]. Mais recentemente, anormalidades neuroinflamatórias e imunológicas foram documentadas em pacientes com transtornos psiquiátricos clássicos.
Moduladores imunológicos periféricos podem induzir sintomas psiquiátricos em modelos animais e humanos [12-19]. Animais saudáveis injetados com IL-1 pró-inflamatória? e as citocinas do fator de necrose tumoral alfa (TNF-?) demonstram comportamento de doença associado ao retraimento social [12]. Em humanos, as injeções de endotoxina em baixas doses desativam o estriado ventral, uma região crítica para o processamento de recompensas, produzindo anedonia, um sintoma depressivo debilitante [14]. Aproximadamente 45% dos pacientes não deprimidos com hepatite C e câncer tratados com IFN-? desenvolver sintomas depressivos associados a níveis séricos elevados de IL-6 [12,15,17,18].
Condições médicas associadas a anormalidades inflamatórias e imunológicas crônicas, incluindo obesidade, diabetes, neoplasias, artrite reumatóide e esclerose múltipla, são fatores de risco para depressão e transtorno bipolar [10,12,13,15,17,18]. O positivo A correlação entre essas condições médicas e doenças psiquiátricas sugere a presença de um processo inflamatório subjacente generalizado que afeta o cérebro entre outros órgãos [10,19,20]. Um estudo baseado na população 30-year mostrou que ter um doença autoimune ou uma hospitalização prévia para infecção grave aumentou o risco de desenvolver esquizofrenia por 29% e 60%, respectivamente [16]. Além disso, o vírus do herpes simple, Toxoplasma gondii, citomegalovírus e gripe durante a gravidez aumentam o risco de desenvolver esquizofrenia [16].
Anormalidades celulares periféricas [21,22] (Tabela 2) e anormalidades imunológicas humorais [13,21-23] são mais prevalentes em pacientes psiquiátricos em relação aos controles saudáveis. Em ambos os estudos piloto (n = 34 pacientes com transtorno depressivo maior (MDD), n = 43 controles saudáveis) e estudos de replicação (n = 36 MDD, n = 43 controles saudáveis), um ensaio sérico compreendendo nove biomarcadores séricos distinguiu indivíduos com TDM de saudáveis controles com sensibilidade de 91.7% e especificidade de 81.3%; biomarcadores significativamente elevados para sintomas neuropsiquiátricos foram as moléculas imunológicas alfa 1 antitripsina, mieloperoxidase e TNF-? receptor II [23].
 Examinamos primeiro a associação entre auto-imunidade e distúrbios neuropsiquiátricos, incluindo: 1) lúpus eritematoso sistêmico (LES) como um protótipo de doença auto-imune sistêmica; 2) encefalites auto-imunes associadas a autoanticorpos séricos anti-sinápticos e de descarboxilase de ácido glutâmico (GAD); e 3) distúrbios auto-imunes neuropsiquiátricos pediátricos associados a infecções estreptocócicas (PANDAS) e distúrbios obsessivo-compulsivos puros (TOC) associados a gânglios anti-basais / autoanticorpos talâmicos. Em seguida, discutimos o papel da inflamação inata / auto-imunidade em distúrbios psiquiátricos clássicos, incluindo MDD, transtorno bipolar (DBP), esquizofrenia e TOC.
Examinamos primeiro a associação entre auto-imunidade e distúrbios neuropsiquiátricos, incluindo: 1) lúpus eritematoso sistêmico (LES) como um protótipo de doença auto-imune sistêmica; 2) encefalites auto-imunes associadas a autoanticorpos séricos anti-sinápticos e de descarboxilase de ácido glutâmico (GAD); e 3) distúrbios auto-imunes neuropsiquiátricos pediátricos associados a infecções estreptocócicas (PANDAS) e distúrbios obsessivo-compulsivos puros (TOC) associados a gânglios anti-basais / autoanticorpos talâmicos. Em seguida, discutimos o papel da inflamação inata / auto-imunidade em distúrbios psiquiátricos clássicos, incluindo MDD, transtorno bipolar (DBP), esquizofrenia e TOC.
Distúrbios neuropsiquiátricos associados à auto-imunidade
Lúpus Eritematoso Sistêmico
Entre 25% a 75% dos pacientes com LES têm envolvimento do sistema nervoso central (SNC), com sintomas psiquiátricos ocorrendo tipicamente nos primeiros dois anos do início da doença. Os sintomas psiquiátricos podem incluir ansiedade, humor e distúrbios psicóticos [97]. A ressonância magnética do cérebro (MRI) é normal em aproximadamente 42% dos casos de LES neuropsiquiátricos [97]. A quebra da microangiopatia e da barreira hematoencefálica (BBB) pode permitir a entrada de autoanticorpos no cérebro [97]. Esses anticorpos incluem anti-P ribossomal (positivo em 90% dos pacientes psicóticos com LES) [1], célula anti-endotelial, anti-gangliósido, anti-dsDNA, subunidades anti-2A / 2B de receptores N-metil-D-aspartato ( NMDAR) e anticorpos antifosfolipídios [97]. Citocinas pró-inflamatórias principalmente IL-6 [97], S100B [97], a molécula de adesão intracelular 1 [97] e a metaloproteinase-9 da matriz [98] também estão elevadas no LES. Manifestações psiquiátricas de LES, doença de Sjo? Gren, síndrome de Susac, vasculite do SNC, doença de Whipple no SNC e doença de Behc? Ets foram revisadas recentemente [1].
Encefalites neuropsiquiátricas auto-imunes associadas ao anti-sináptico sérico e à descarboxilase do ácido glutâmico
Autoanticorpos
As encefalites autoimunes são caracterizadas por um início agudo de convulsões do lobo temporal, características psiquiátricas e déficits cognitivos [2,3,99-108]. A fisiopatologia é tipicamente mediada por autoanticorpos direcionados a autoantígenos sinápticos ou intracelulares em associação com uma origem paraneoplástica ou não paraneoplásica [3]. Os autoanticorpos anti-sinápticos têm como alvo as subunidades NR1 do NMDAR [100,108,109], complexos de canal de potássio controlado por voltagem (VGKC) (subunidade Kv1, glioma rico em leucina inativado (LGI1) e proteína associada à contactina 2 (CASPR2)) [101,102,106], GluR1 e Subunidades GluR2 do receptor do ácido amino-3-hidroxi-5-metil-l-4-isoxazolpropiônico (AMPAR) [6,110,111] e subunidades B1 dos receptores B do ácido? -Aminobutírico (GABABR) [3,99,103]. Os autoanticorpos anti-intracelulares têm como alvo os autoantígenos onconeuronal e GAD-65 [2,3].
A inflamação associada aos autoanticorpos anti-sinápticos, particularmente os autoanticorpos NMDAR, tipicamente é muito mais suave do que a associada a autoanticorpos GAD ou autoanticorpos anti-neuronais relacionados a distúrbios auto-imunes sistêmicos ou síndromes paraneoplásicas [2,107].
Embora os sintomas neurológicos eventualmente surjam, as manifestações psiquiátricas, que vão desde a ansiedade [2,3] até a psicose imitando a esquizofrenia [2-6], podem inicialmente dominar ou preceder características neurológicas. Até dois terços dos pacientes com encefalite auto-imune NMDAR, inicialmente presente em serviços psiquiátricos [5]. As encefalites autoimunes mediadas por anticorpos anti-sinápticos devem ser consideradas no diferencial de psicose aguda [2-6]. As apresentações psiquiátricas podem incluir a análise cerebral normal de MRI e líquido cefalorraquidiano (LCR), sem encefalopatia ou convulsões [2,3,5,6,107]. Relatamos um caso de autoanticorpos GAD soropositivos associados à neuroinflamação comprovada por biópsia, apesar das análises normais de MRI e CSF no cérebro, onde o paciente apresentou psicose isolada diagnosticada como esquizofrenia por Diagnóstico e Estatística de Distúrbios Mentais, 4th Edition (DSM-IV) [2]. Além disso, as encefalites auto-imunes seronegativas também podem apresentar distúrbios neuropsiquiátricos proeminentes, tornando o diagnóstico mais evasivo [107,112,113]. As características psiquiátricas e neurológicas associadas aos autoanticorpos anti-sinápticos e GAD estão resumidas na Tabela 1 [1-6,99-108,114].
Os autoanticorpos anti-sinápticos séricos e GAD podem ocorrer em pacientes com transtornos psiquiátricos puros [2,4,5,112,115-121]. Em uma coorte prospectiva de 29 indivíduos que preencheram os critérios do DSM-IV para esquizofrenia, autoanticorpos anti-NMDAR séricos foram encontrados em três indivíduos, e autoanticorpos anti-VGKC-complexo foram encontrados em um indivíduo [5]. Usando técnicas mais sensíveis para detectar autoanticorpos NR1 da imunoglobulina G (IgG) em 100 pacientes com esquizofrenia definitiva, nenhum autoanticorpo foi identificado [122]. No entanto, este estudo não avaliou autoanticorpos direcionados à subunidade NR2 de NMDAR. Outros estudos relataram chances significativamente aumentadas de níveis de anticorpo NR90 elevados (? 2º percentil) níveis de anticorpos NR2.78 (razão de chances (OR) 95, intervalo de confiança de 1.26% (IC) 6.14 a 0.012, P = 43) entre indivíduos com mania aguda ( n = 116), mas não na mania crônica ou esquizofrenia [XNUMX].
PANDAS e transtorno obsessivo-compulsivo puro associado a autoanticorpos anti-gânglios basais / talâmicos
O TOC freqüentemente complica distúrbios neurológicos envolvendo os gânglios da base, incluindo a coreia de Sydenham, a doença de Huntington e a doença de Parkinson. Anticorpos anti-gânglios basais estão implicados na coréia de Sydenham [123]. PANDAS é caracterizado por exacerbações agudas dos sintomas de TOC e / ou tiques motores / fônicos após uma infecção prodrômica por estreptococos do grupo A? -Hemolítico. Acredita-se que a fisiopatologia envolva reatividade cruzada entre anticorpos anti-estreptocócicos e proteínas dos gânglios da base [124]. A sobreposição clínica entre o PANDAS e o TOC puro sugere um mecanismo etiológico comum [125].
Entre uma coorte aleatória de 21 pacientes com TOC puro, 91.3% tinham gânglios anti-basais no LCR (P <0.05) e autoanticorpos anti-talâmicos (P <0.005) a 43 kDa [88], paralelamente a anormalidades funcionais no córtex-estriado-tálamo - Circuito cortico de sujeitos com TOC [84]. Outro estudo documentou que 42% (n = 21) dos indivíduos pediátricos e adolescentes com TOC tinham autoanticorpos anti-gânglios da base séricos em 40, 45 e 60 kDa em comparação com 2% a 10% dos controles (P = 0.001) [7]. Auto-anticorpos anti-gânglios basais foram detectados no soro de 64% dos indivíduos PANDAS (n = 14), em comparação com apenas 9% (n = 2) dos controles estreptocócicos / OCD-negativos (P <0.001) [126]. Um estudo não encontrou nenhuma diferença entre a prevalência de autoanticorpos anti-gânglios basais no TOC (5.4%, n = 4) versus controles com TDM (0%) [127]; entretanto, uma limitação foi o uso aleatório de córtex de rato e gânglios basais e córtex bovinos que pode ter limitado a identificação de casos soropositivos.
Os autoantígenos dos gânglios basais são aldolase C (40 kDa), enolase neuronal-específica / não neuronal (dupleto de 45 kDa) e piruvato quinase M1 (60 kDa) enzimas glicolíticas neuronaisenvolvidas na neurotransmissão, metabolismo neuronal
Página 3 de 24 e sinalização celular [128]. Estas enzimas exibem homologia estrutural substancial às proteínas estreptocócicas [129]. O último estudo (96 OCD, 33 MDD, 17 esquizofrênico) testou o soro do paciente contra piruvato quinase, aldolase C e enolase, especificamente; uma proporção maior de indivíduos com TOC foi seropositiva em relação aos controles (19.8% (n = 19) versus 4% [n = 2], P = 0.012) [130].
No entanto, no mesmo estudo, apenas um dos indivíduos XOUMX seropositivos do TOC também apresentou anticorpos anti-estreptolisina O positivos, sugerindo que, no TOC puro, a seronegatividade do anticorpo anti-estreptolisina O não exclui a presença de autoanticorpos anti-ganglio anti-basal .
No TOC puro, a sero-positividade para os gânglios anti-basal / anticorpos talâmicos está associada a níveis aumentados de glicina CSF (P = 0.03) [88], sugerindo que esses anticorpos contribuem para a hiperglutamatergia observada no TOC [84,88,131]. A melhora do TOC provocado por infecção com terapias imunes suporta a patogenicidade desses auto-anticorpos [132]. Um grande estudo NIH avaliando a eficácia da imunoglobulina intravenosa (IVIG) para crianças com TOC de início agudo e anticorpos anti estreptocócicos está em andamento (ClinicalTrials.gov: NCT01281969). No entanto, a descoberta de níveis ligeiramente superiores de glutamato de LCR em pacientes com TOC com órgãos anti-ganglio / tálamo contra-basal negativos de CSF em comparação com aqueles com anticorpos positivos de LCR sugere que mecanismos não-imunológicos podem desempenhar papel no TOC [84]. Outros mecanismos, incluindo a inflamação mediada por citocinas (Tabela 2), também são hipóteses.
Distúrbios psiquiátricos associados à inflamação inata
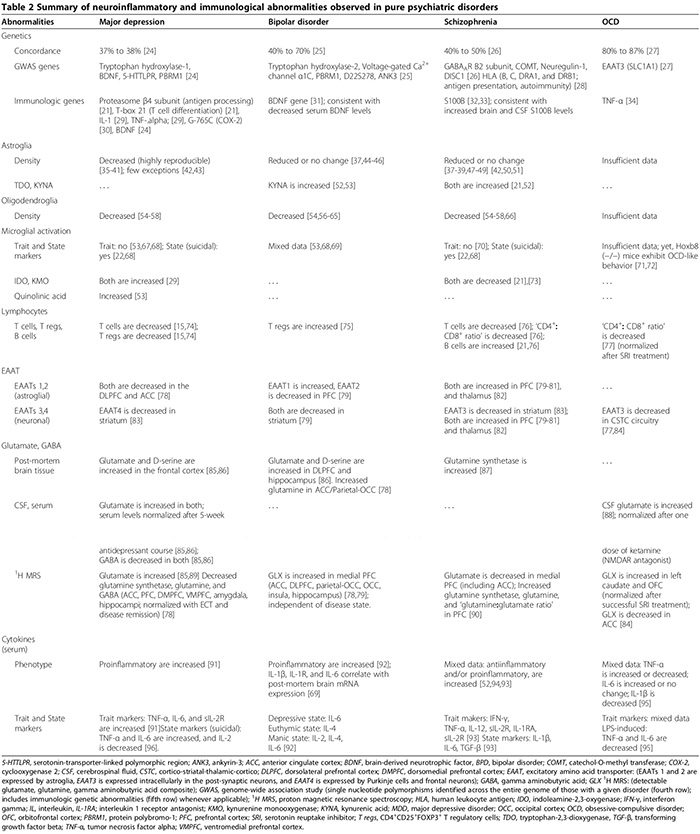
Distúrbios de inflamação / autoimunidade inata ocorrem em alguns pacientes com distúrbios psiquiátricos clássicos. Discutimos anormalidades do SNC relacionadas à inflamação inata incluindo patologia glial, níveis elevados de citocinas, ativação da ciclooxigenase, desregulação do glutamato, aumento dos níveis de S100B, aumento do estresse oxidativo e disfunção BBB em MDD, DBP, esquizofrenia e TOC. Também descrevemos como a inflamação inata pode estar mecanicamente ligada às anormalidades monoaminérgicas e glutamatérgicas tradicionais relatadas nesses distúrbios (Figuras 1 e 2). O papel terapêutico dos agentes antiinflamatórios em transtornos psiquiátricos também é revisado.
 Histopatologia Astroglial e Oligodendroglial
Histopatologia Astroglial e Oligodendroglial
Astroglia e oligodendroglia são essenciais para neural Metabólico homeostase, comportamento e funções cognitivas superiores [54-56,133-136]. A astroglia quiescente normal fornece energia e suporte trófico aos neurônios, regula a neurotransmissão sináptica (Figura 2), a sinaptogênese, o fluxo sanguíneo cerebral e mantém a integridade da BHE [134,136,137]. Oligodendróglias maduras fornecem energia e suporte trófico aos neurônios e mantêm a integridade BBB e regulam o reparo axonal e mielinização de tratos de matéria branca que fornecem conectividade inter e intra-hemisférica [54-56]. Tanto astroglia quanto oligodendroglia produzem citocinas anti-inflamatórias que podem reduzir a inflamação prejudicial [52,55].
Na MDD, a perda astroglial é um achado pós-mortem consistente em áreas funcionalmente relevantes, incluindo córtex cingulado anterior, córtex pré-frontal, amígdala e matéria branca [35-38,42-46,55,138-147], com poucas exceções [42,43]. Os estudos pós-mortem revelaram uma densidade astroglial positiva de proteína glial fibrilante reduzida (GFAP), principalmente no córtex pré-frontal [37,38] e amígdala [36]. Uma grande análise proteômica de córtices frontais de pacientes deprimidos mostrou reduções significativas em três isoformas de GFAP [39]. Embora em um estudo que não relatou perda glial significativa, a análise de subgrupos revelou uma diminuição significativa (75%) na densidade astroglial positiva de GFAP em indivíduos com idade menor que 45 anos de idade [35]. Um estudo morfométrico também não mostrou alterações na densidade glial nos cérebros MDD tardios [148]. Nós levantamos a hipótese de que a ausência aparente de perda astroglial em pacientes idosos de MDD pode refletir a astrogliose secundária [35] que está associada a idade mais avançada [42,50] em vez de um verdadeiro negativo.
Os estudos em animais são consistentes com estudos em humanos que mostram perda astroglial em MDD. Ratos Wistar-Kyoto conhecidos por exibirem comportamentos semelhantes aos depressivos revelaram densidade astroglial reduzida nas mesmas áreas observadas em humanos [40]. A administração do agente tóxico astroglial, ácido L-alfa-aminoadípico, induz sintomas semelhantes aos da depressão em ratos, sugerindo que a perda astroglial é patogênica no TDM [41].
Estudos pós-mortem de sujeitos MDD documentaram a densidade oligodendroglial reduzida no córtex pré-frontal e amígdala [54-57,66], que pode correlacionar-se com as alterações focais focais de MRI específicas ocasionalmente observadas em alguns pacientes com MDD [57]. No entanto, as anormalidades microvasculares também podem contribuir para essas mudanças [57].
Na BPD, alguns estudos demonstram perda glial significativa [138,143,149,150], enquanto outros não [37,44-46]. Essas descobertas inconsistentes podem resultar da falta de controle para o tratamento 1 com estabilizadores de humor, pois a análise pós-hoc relatada por alguns estudos mostrou redução significativa na perda glial somente após o controle do tratamento com ácido lítio e valproico [46]; 2) formas familiares de DBP, uma vez que a perda glial é particularmente proeminente entre os pacientes com DBP com forte história familiar [143]; e / ou, 3) o estado predominante de depressão versus mania, como a perda glial é freqüente em MDD [35-38,42-46,55,138-147]. Se a astroglia ou a oligodendroglia representam a maioria da perda glial não está clara; enquanto a análise proteômica revelou uma diminuição significativa em uma isoforma astroglial de GFAP [39], vários outros estudos pós-mortem descobriram inalterados [36,37] ou redução da expressão astroglial positiva no GFAP no córtex orbitrofrontal [47] ou redução da densidade oligodendroglial [54- 56,58,59].
Na esquizofrenia, a perda astroglial é um achado inconsistente [48,150]. Enquanto alguns estudos não mostraram perda significativa de astroglial [42,50,51], vários outros encontraram a densidade astroglial reduzida [37,38,43,44,48,49,151] e reduções significativas em duas isoformas de GFAP [39]. Achados inconsistentes podem resultar de: 1) comorbidade MDD, que geralmente está associada à perda glial; 2) variação da idade, uma vez que os pacientes idosos aumentaram astroglia positiva ao GFAP [35,42,50]; 3) regional [150] e variabilidade da camada cortical [48]; 4) com fármacos antipsicóticos, uma vez que estudos experimentais mostram redução [152] e aumento da densidade astroglial [153] relacionada ao tratamento antipsicótico crônico [70]; e 5) estado da doença (por exemplo, comportamento suicida versus não suicida) [154]. Os estudos post mortem documentaram perda oligodendroglial [54,56,60-65,148,155,156], particularmente no córtex pré-frontal, córtex cingulado anterior e hipocampo [148]. O exame ultra-estrutural da região pré-frontal mostrou fibras anormalmente mielinizadas em matéria cinza e branca; Tanto a idade quanto a duração da doença foram positivamente correlacionadas com as anormalidades da substância branca [157].
Em contraste com distúrbios neurodegenerativos que são comumente associados à proliferação astroglial [136], os distúrbios psiquiátricos são, em vez disso, associados à densidade astroglial reduzida ou inalterada [138]. A falta de aumento da densidade glial em distúrbios psiquiátricos de início precoce [44,138] pode refletir a taxa mais lenta de progressão degenerativa em doenças psiquiátricas [138].
Postulamos que as alterações degenerativas associadas a transtornos psiquiátricos são mais sutis e não graves o suficiente para provocar fatores de transcrição intracelular astroglial que regulam positivamente a astrogliose, incluindo transdutor de sinal ativador de transcrição 3 e fator nuclear kappa B (NF-? B) [136].
Embora a maioria dos estudos pós mortem tenha se concentrado na alteração da densidade glial em MDD, DBP e esquizofrenia, outros descreveram a alteração da morfologia das células gliais, com achados mistos. Em MDD e BPD, o tamanho glial é aumentado ou inalterado [55]. Um estudo encontrou redução do tamanho glial na DBP e esquizofrenia, mas não em MDD [43]. Um estudo post-mortem de pacientes deprimidos que cometeu suicídio encontrou aumento do tamanho astroglial na substância branca cingulada anterior, mas não no córtex [158]. Um estudo em indivíduos esquizofrênicos encontrou redução acentuada do tamanho astroglial na camada V do córtex pré-frontal dorsolateral, apesar de a densidade astroglial ser o dobro dos controles na mesma camada [48]. Os resultados mistos podem refletir parcialmente estudos anteriores de alterações gliais em doenças psiquiátricas que não especificaram astroglia versus oligodendroglia [148].
A perda glial em doenças psiquiátricas pode contribuir para a neuroinflamação por meio de vários mecanismos, incluindo níveis anormais de citocinas (consulte a seção Citocinas), metabolismo desregulado do glutamato (consulte a seção Glutamato), proteína S100B elevada (consulte a seção S100B), e função BBB alterada (ver seção de barreira do cérebro sangüíneo), resultando em cognição e comportamento prejudicados [44,45,54,133,159].
Histopatologia microglial
A microglia é a célula imune residente do SNC. Eles fornecem vigilância imunológica contínua e regulam a poda sináptica do desenvolvimento [160,161]. A lesão do SNC transforma a microglia de apoio ramificada em células amebitas fagocíticas fagocíticas alongadas activadas e em forma de bastonete que proliferam e migram para o local da lesão ao longo de gradientes quimiotáticos (isto é, ativação e proliferação microvasculares (MAP)) [161]. As células microgliais humanas expressam NMDARs que podem mediar MAP levando a lesão neuronal [162].
Em MDD, BPD e esquizofrenia, os resultados dos estudos pós-mortem que investigam a presença de MAP são misturados. Estudos pós-mortem revelaram MAP elevado em apenas um em cada cinco indivíduos MDD [67]. Em alguns pacientes com transtorno de DBP, o aumento da microglia DR-positiva de antígeno leucocitário humano mostrando processos engrossados foi documentado no córtex frontal [69]. Na esquizofrenia, enquanto alguns estudos relataram MAP elevado em relação aos controles, outros não mostraram diferença entre os grupos [22,67,70]. Em um estudo post-mortem avaliando MAP em MDD e BPD; A densidade de células microgliais positivas ao ácido quinolínico foi aumentada no córtex cingulado anterior subgenial e no córtex intermediário anterior de pacientes com MDD e DBP que se suicidaram em relação aos controles [53]. A análise pós-hoc revelou que o MAP aumentado era exclusivamente atribuível à MDD e não à DBP, uma vez que a imunocoloração microglial positiva em indivíduos com MDD era significativamente maior do que no subgrupo de DBP nos córtex subgeneral anterior e cortimentos midginados e, como a densidade de microglia foi semelhante em BPD e grupos de controle [53]. Um estudo comparando os três distúrbios (nove MDD, cinco BPD, catorze esquizofrenia, dez controles saudáveis) não demonstrou diferença significativa na densidade microglial nos quatro grupos [68].
Estes resultados mistos podem ser atribuídos a marcadores imunológicos microgliais variáveis utilizados entre diferentes estudos [70] e / ou a falta de controle para a gravidade da doença [22,53,68]. Notavelmente, três estudos pós-mortem de MDD e indivíduos esquizofrênicos documentaram uma forte correlação positiva entre MAP e suicídio no córtex cingulado anterior e tálamo mediodórtrico, dependendo do diagnóstico psiquiátrico [22,53,68]. Assim, MAP pode ser um estado em vez de um marcador de traços para MDD e esquizofrenia.
No TOC, modelos animais sugerem que a disfunção e a redução de certos fenótipos microgliais, como aqueles que expressam o gene Hoxb8, que codifica o fator de transcrição do homeobox, podem causar um comportamento semelhante ao TOC [71,72].
Os ratos knockout Hoxb8 exibem comportamento de higiene excessivo e ansiedade em associação com a densidade microglial reduzida [71,72]. Este comportamento de higiene excessiva se assemelha às características comportamentais do TOC humano. A injeção de Hoxb8 em ratos adultos com nocaute Hoxb8 inverte a perda microglial e restaura o comportamento normal [71,72]. O papel desses fenótipos microgliais específicos no TOC humano não está claro.
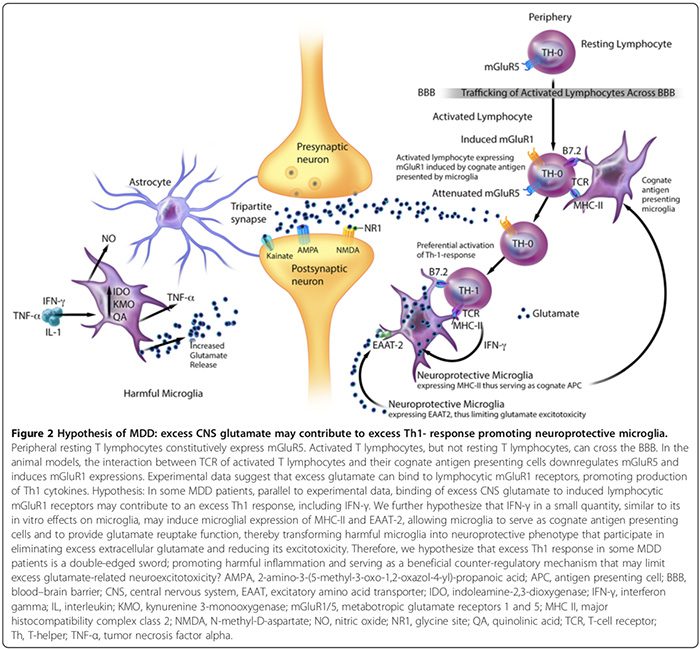
Dados experimentais sugerem que o MAP compreende fenótipos prejudiciais e neuroprotetivos distintivos (Figura 2). A microglia prejudicial não expressa o complexo de histocompatibilidade II (MHC-II) e, portanto, não pode atuar como células apresentadoras de antígeno (APC) [163,164]; eles promovem efeitos deletérios [17,69,165] através da produção de citocinas pró-inflamatórias, sinalização de sintetização de óxido nítrico [17,166], promovendo a expressão Glial e BBB-pericyte / ciclooxigenase endotelial-2 (COX-2) [167], induzindo secreção astroglial S100B (ver seção S100B) , e liberação de glutamato microglial [17,136,168,169]. A microglia prejudicial também secreta a prostaglandina E-2 (PGE-2) que promove a produção de citocinas pró-inflamatórias, que por sua vez aumenta os níveis de PGE-2 em um ciclo de avanço [29]. Além disso, PGE-2 estimula a expressão COX-2, que medeia a conversão de ácido araquidônico em PGE-2, configurando outro ciclo de avanço [29].
A microglia neuroprotetiva em contraste pode: 1) expressar MHC-II in vivo e in vitro [163,166] e atuar como APC cognato (Figura 2) [163,164,166]; 2) facilitam a cura e limitam a lesão neuronal promovendo a secreção de citocinas anti-inflamatórias [17], fator neurotrófico derivado do cérebro [17] e fator de crescimento similar a insulina-1 [166]; e 3) expressam o transdutor de aminoácidos excitatório-2 (EAAT2) que elimina o excesso de glutamato extracelular [163,166] e promove a auto-imunidade linfocítica T neuroprotetiva (Figura 2) [163,164]. No entanto, são necessários mais estudos para confirmar o papel contributivo da microglia neuroprotetiva para distúrbios neuropsiquiátricos em seres humanos.
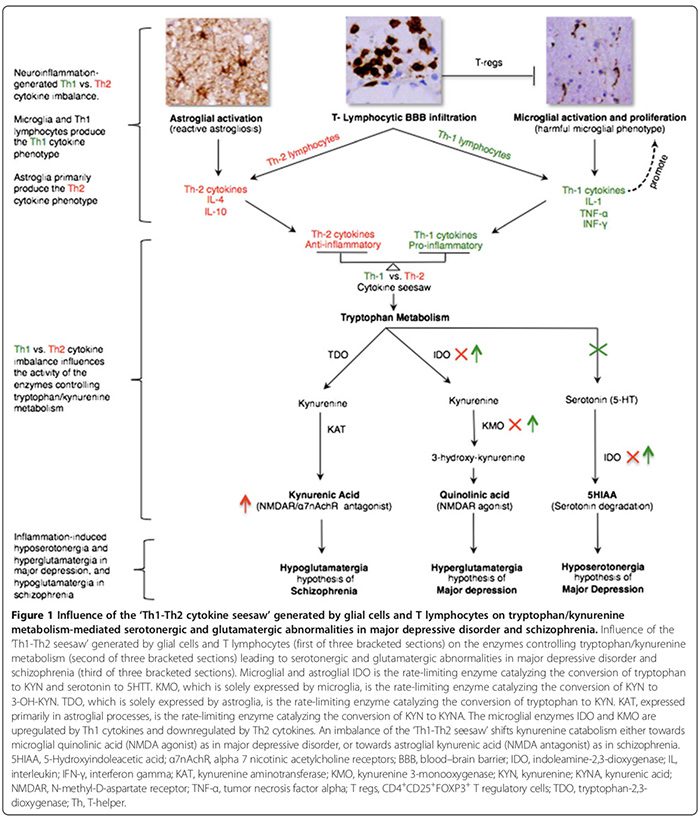
In vitro estudos em animais sugerem que a proporção de microglia prejudicial versus neuroprotetora pode ser influenciada pelo efeito líquido de mecanismos contra-reguladores inflamatórios [15,74,164,166]. Esses mecanismos incluem o número de células T reguladoras neuroprotetoras CD4 + CD25 + FOXP3 + ((T regs) Figura 1) [15,74,164,166] e níveis de citocinas cerebrais; baixo IFN-? níveis podem promover microglia neuroprotetora (Figura 2) [166], enquanto níveis elevados podem promover o fenótipo prejudicial [166].
O papel das citocinas
As citocinas pró-inflamatórias incluem IL-1 ?, IL-2, IL-6, TNF-? e IFN- ?. Eles são secretados principalmente pela micro-glia, linfócitos Th1 e monócitos / macrófagos do fenótipo M1 (Figura 1) [15,170]. Eles promovem inflamações prejudiciais. As citocinas antiinflamatórias incluem IL-4, IL-5 e IL-10. Eles são secretados principalmente pela astroglia, Linfócitos Th2, T regs e monócitos de fenótipo M2 / macrófagos [15,52,74]. Eles podem limitar a inflamação nociva [15,74], convertendo o tipo de fenômeno proinflamatório M1 para o fenótipo antiinflamatório M2 benéfico [15] e potencialmente promovendo o fenótipo microglial neuroprotetivo [15,17,74,163,166]. O papel das citocinas pró-inflamatórias / anti-inflamatórias em distúrbios psiquiátricos é suportado por várias linhas de evidência (Figura 1, Tabela 2) [15,17,29,52,74].
No MDD, a meta-análise mais recente (29 estudos, 822 MDD, 726 controles saudáveis) de citocinas pró-inflamatórias séricas confirmou que o receptor solúvel de IL-2, IL-6 e TNF-? os níveis estão aumentados em MDD (marcadores de traço) [91], enquanto IL-1 ?, IL-2, IL-4, IL-8 e IL-10, não são estatisticamente diferentes dos controles [91]. Em um estudo primário de citocinas comparando subgrupos de TDM (47 TDM suicida, 17 TDM não suicida, 16 controles de saúde), ambos os soros IL-6 e TNF-? foram significativamente mais altos, enquanto os níveis de IL-2 foram significativamente mais baixos em indivíduos com TDM que cometeram suicídio em relação a ambos os outros grupos [96]. Esse achado sugere que IL-6 e TNF-? também são marcadores de estado de MDD [96]. A diminuição dos níveis séricos de IL-2 associados ao comportamento suicida agudo pode refletir o aumento da ligação ao seu receptor regulado positivamente no cérebro; paralelamente à meta-análise mencionada acima mostrando aumento do receptor solúvel de IL-2 em MDD [91]. Estudos que investigam a significância clínica das citocinas no TDM mostraram que os níveis séricos de citocinas são elevados durante episódios depressivos agudos [171,172] e normalizados após o sucesso, mas não falha, do tratamento com antidepressivos [17] e terapia eletroconvulsiva [29]; esses achados sugerem um possível papel patogênico para as citocinas.
No DBP, as alterações das citocinas séricas foram resumidas em uma revisão recente; TNF- ?, IL-6 e IL-8 são elevados durante as fases maníaca e depressiva, enquanto IL-2, IL-4 e IL-6 estão elevados durante a mania [92]. Outros estudos mostraram que o soro IL-1? e os níveis do receptor de IL-1 não são estatisticamente diferentes dos controles saudáveis [92], embora estudos de tecidos tenham documentado níveis aumentados de IL-1? e receptor de IL-1 no córtex frontal da DBP [69].
Na esquizofrenia, os resultados dos estudos que investigam as anormalidades das citocinas são conflitantes (Tabela 2). Enquanto alguns estudos encontraram uma diminuição sérica pró-inflamatória (IL-2, IFN-?) E aumento sérico e citocinas antiinflamatórias (IL-10) do LCR [52], outros encontraram citocinas séricas pró-inflamatórias e antiinflamatórias elevadas, com uma dominância do tipo pró-inflamatório [22,173,174 ] Uma meta-análise de citocinas (62 estudos, 2,298 esquizofrenia, 858 controles saudáveis) mostrou níveis aumentados de antagonista de IL-1R, sIL-2R e IL-6 [174]. No entanto, este estudo não levou em consideração o uso de antipsicóticos, que se acredita aumentar a produção de citocinas pró-inflamatórias [52]. Uma meta-análise de citocinas mais recente (40 estudos, 2,572 esquizofrênicos, 4,401 controles) responsáveis pelos antipsicóticos, descobriram que TNF- ?, IFN- ?, IL-12 e sIL-2R estão consistentemente elevados na esquizofrenia crônica independente da atividade da doença (marcadores de traço), enquanto IL-1 ?, IL-6 e o fator transformador de crescimento beta correlaciona-se positivamente com a atividade da doença (marcadores estaduais) [173]. As culturas de células mononucleares do sangue periférico (PBMC) obtidas de pacientes esquizofrênicos produziram níveis mais elevados de IL-8 e IL-1? espontaneamente, bem como após estimulação por LPS, sugerindo um papel para monócitos / macrófagos ativados na patologia da esquizofrenia [175].
No TOC, os resultados de uma pesquisa aleatória de soros e citocinas CSF, e estudos de PBMC estimulados por LPS, são inconsistentes [93-95,176-179]. Existe uma correlação entre o TOC e um polimorfismo funcional na região promotora do TNF-? gene [34], embora estudos de baixa potência não tenham confirmado esta associação [180]. Portanto, os resultados mistos de estudos que documentam o aumento ou a diminuição do TNF-? os níveis de citocinas [93,176-178] podem refletir sua inclusão variável do subconjunto de indivíduos com TOC com este polimorfismo específico em suas coortes.
Polarização da resposta a citocinas na grande depressão e esquizofrenia
Os fenótipos de resposta às citocinas são classificados como Th1 pró-inflamatório (IL-2, IFN-?) Ou Th2 antiinflamatório (IL-4, IL-5, IL-10) de acordo com as funções imunológicas que regulam. Enquanto as citocinas Th1 regulam a imunidade mediada por células dirigida contra antígenos intracelulares, as citocinas Th2 regulam a imunidade humoral dirigida contra antígenos extracelulares [29,52]. As citocinas Th1 são produzidas por linfócitos Th1 e monócitos M1, enquanto as citocinas Th2 são produzidas por linfócitos Th2 e monócitos M2 [29,52]. No cérebro, a microglia secretam predominantemente citocinas Th1, enquanto a astroglia secretam predominantemente citocinas Th2 [29,52]. A proporção recíproca de citocinas Th1: Th2, doravante Th1-Th2 gangorra, é influenciada pela proporção de microglia ativada (excesso de Th1) para astroglia (excesso de Th2) e a interação entre células T ativadas e níveis excessivos de glutamato do SNC que hipotetizamos para favorecer a resposta Th1 (Figura 2) [29,163,166].
O desequilíbrio de gangorra Th1-Th2 pode influenciar o metabolismo do triptofano, alterando suas enzimas [21,52], deslocando o catabolismo de triptofano em direção ao kinurenina (KYN) e ao catabolismo de KYN para qualquer um dos seus dois metabolitos descendentes; ácido quinolínico de microglia que é mediado por medição de Th1 ou ácido kinurênico astroglial (KYNA) (Figura 1) que é mediada por Th2 mediada por [21,29,170].
As enzimas do metabolismo do triptofano afetadas pela gangorra Th1-Th2 incluem (Figura 1): indoleamina 2,3-dioxigenase (IDO) expressa pela microglia e astroglia, as enzimas limitantes da taxa que medeiam a conversão de triptofano em KYN e serotonina em 5- ácido hidroxiindolacético [21,29]. A quinurenina 3-monooxigenase (KMO), expressa apenas pela microglia, é a enzima limitadora da taxa que converte KYN em 3-hidroxiquinurenina (3-OH-KYN), que é posteriormente metabolizada em ácido quinolínico [21,29]. Triptofano-2,3-dioxigenase (TDO), expresso exclusivamente pela astroglia, é a enzima limitadora da taxa que converte triptofano para KYN [21,29]. Kynurenine aminotransferase (KAT), expressa principalmente em processos astrogliais, é a enzima limitante de taxa que medeia a conversão de KYN para KYNA [21,29].
As citocinas Th1 ativam IDO e KMO microglial, mudando o catabolismo KYN microglial para quinolínico síntese de ácido (agonista de NMDAR), enquanto que as citocinas Th2 ativam o IDO e o KMO microgliais, deslocando o catabolismo astroglial KYN para síntese KYNA (antagonista NMDAR mediada por TDO e KAT) (Figura 1) [21,29].
Os imunofenótipos predominantes Th1 e Th2 foram propostos para TDM e esquizofrenia, respectivamente, com base em padrões de citocinas periféricas, em vez de no SNC [52,173]. Acreditamos que os padrões de citocinas periféricas são marcadores substitutos não confiáveis daqueles no SNC. De fato, os níveis periféricos de citocinas podem ser influenciados por muitas variáveis extra-SNC, que não são consistentemente controladas em vários dos estudos de citocinas periféricas, incluindo: 1) idade, índice de massa corporal, medicamentos psicotrópicos, tabagismo, estresse e flutuações circadianas; 2) a influência de atividade / estado da doença na produção de síntese de citocinas selecionadas [95,173]; e 3) os efeitos dos agentes psicotrópicos na produção de citocinas [52]. A meia-vida curta e a rápida renovação das citocinas séricas [181] (por exemplo, 18 minutos para TNF-? [182] versus 60 minutos para IL-10 [183]) podem limitar ainda mais a confiabilidade da interpretação de seus níveis medidos a partir de amostragem aleatória de soros.
No TDM, há um consenso de que uma resposta imunofenotípica Th1 pró-inflamatória predomina (Tabela 2) [17,29]. Altos níveis de ácido quinolínico em cérebros MDD post-mortem [53], sugerem a presença de uma resposta Th1 regulada positivamente (Figura 1) [21,29]. O ácido quinolínico do SNC elevado pode promover a apoptose mediada pelo influxo de cálcio da astroglia humana [184], o que, hipoteticamente, pode embotar oresposta Th2 derivada de astroglia [29], inclinando Th1 contra saldo de balanço Th2 em favor da resposta microglial Th1. A hiposserotonergia do SNS [29] adiciona suporte adicional a uma resposta Th1 em excesso, que mostra reduzir a síntese de serotonina do SNC [185] e aumentar sua degradação (Figura 1) [21,29].
A hiperglutamatergia do SNC também pode contribuir para uma resposta Th1 em excesso no cérebro (Figura 2). Um estudo in vitro sugere que os linfócitos T em repouso periféricos expressam constitutivamente o receptor de glutamato metabotrópico 5 (mGluR5) [164], cuja ligação ao glutamato inibe a liberação linfocítica de IL-6, minimizando assim a proliferação de células efectoras T auto-reativas [164]. Os linfócitos T ativados, mas não os linfócitos T de repouso, podem atravessar o BBB [37].
Dados experimentais sugerem que a interação entre os receptores de células T de linfócitos T ativados e suas células apresentadoras de antígenos cognatos pode diminuir a regulação de mGluR5 e induzir expressões de mGluR1 [164]. Em modelos animais, a ligação do excesso de glutamato aos receptores linfocíticos mGluR1 promove a produção de citocinas Th1, incluindo IFN-? [164].
Nossa hipótese é que em alguns pacientes com TDM, paralelamente aos dados experimentais [164], a ligação do excesso de glutamato do SNC a receptores mGluR1 linfocíticos induzidos pode contribuir para um excesso de resposta Th1, incluindo IFN-? (Figura 2). Especulamos que IFN-? em uma pequena quantidade, semelhante aos seus efeitos in vitro na microglia [166], pode induzir a expressão microglial de MHC-II e EAAT2 [163,166], permitindo que a microglia sirva como células apresentadoras de antígeno cognato e forneça a função de recaptação de glutamato [163,164,166], transformando assim microglia prejudicial em fenótipo neuroprotetor [163,166] que participa na eliminação do excesso de glutamato extracelular [163,164,166]. Portanto, também levantamos a hipótese de que o excesso de resposta Th1 em subgrupos de pacientes com TDM é uma faca de dois gumes, promovendo inflamação prejudicial e servindo como um mecanismo contrarregulatório benéfico que pode limitar o excesso de neuroexcitotoxicidade relacionada ao glutamato (Figura 2).
Na esquizofrenia, enquanto alguns estudos de citocinas periféricas sugerem a predominância de um imunofenótipo / resposta antiinflamatório Th2 [52], outros refutam isso [173,174]. No entanto, concordamos com os autores que levantaram a hipótese de que a resposta Th2 é o fenótipo dominante na esquizofrenia [52]. Níveis elevados de cérebro, LCR e soro de KYNA [21,52] sugerem regulação negativa de IDO micro-glial e KMO, que é uma função da resposta Th2 que muda o catabolismo KYN astroglial em direção à síntese de KYNA (Figura 1) [21,52]. A atividade reduzida de KMO e a expressão de mRNA de KMO em cérebros esquizofrênicos post-mortem [73] são consistentes com o excesso de resposta Th2 (Figura 1). Aumento da prevalência de anormalidades da imunidade humoral mediada por Th2 em subgrupos de pacientes com esquizofrenia como evidenciado pelo aumento da contagem de células B [21,76], aumento a produção de autoanticorpos incluindo anticorpos antivirais [76] e aumento da imunoglobulina E [52] adiciona suporte adicional à hipótese de dominância da resposta Th2.
Neuroinflamação e desregulação do glutamato do SNC
O glutamato medeia cognição e comportamento [186]. Os níveis de glutamato sintético são regulados por EAATs gliais e neuronais dependentes de sódio de alta afinidade, nomeadamente o sistema XAG responsável pela reabsorção de glutamato / liberação de aspartato [137,164] e o sistema antiderrapato astroglial de glutamato / cistina independente de sódio (Xc-) responsável pela libertação de glutamato / recaptação de cistina [164]. Astroglial EAAT1 e EAAT2 fornecem mais de 90% de reabastecimento de glutamato [79].
Neuroinflamação pode alterar o metabolismo do glutamato e a função de seus transportadores [15,29,187,188], produzindo deficiências cognitivas, comportamentais e psiquiátricas [15,21,29,79,186,188,189]. Anormalidades da função / expressão de EAATs e do metabolismo do glutamato em MDD, DBP, esquizofrenia e TOC estão resumidas na Tabela 2.
Na MDD, há evidências de hiperglutamatergia cortical (Tabela 2). Os níveis de glutamato cortical correlacionaram-se positivamente com a gravidade dos sintomas depressivos, e um curso de cinco semanas de antidepressivos diminuiu as concentrações séricas de glutamato [85,86]. Uma única dose de cetamina, um potente antagonista de NMDAR, pode reverter MDD refratário por uma semana [17,21,29,85]. O excesso de níveis de glutamato do SNC pode induzir inflamação mediada por neurotoxicidade [163,164,188], incluindo uma resposta proinflamatória Th1 (Figura 2) [164].
Evidências in vitro limitadas sugerem que a inflamação / citocinas pró-inflamatórias podem aumentar os níveis de glutamato do SNC [188] em um ciclo de alimentação por meio de vários mecanismos potenciais: 1) as citocinas pró-inflamatórias podem inibir [15,17,168] e reverter [45,137] o glutamato mediado por EAAT astroglial função de recaptação; 2) citocinas pró-inflamatórias podem aumentar a síntese de ácido quinolínico microglial [53], que foi experimentalmente mostrado para promover a liberação de glutamato sinaptossomal [15,17,29,190]; 3) aumento da COX-2 / PGE-2 e do TNF-? os níveis podem induzir o influxo de cálcio [137], que, com base em dados in vitro, pode aumentar a liberação de glutamato astroglial e D-serina [191]; e 4) a microglia ativada pode expressar o excesso de sistemas antiporter Xc que medeiam a liberação de glutamato [164,192].
Na esquizofrenia, são encontradas hipoglutamatergia cortical pré-frontal [87,90,193,194] (Tabela 2) e funcionalidade NMDAR reduzida [5]. A recente meta-análise de espectroscopia de ressonância magnética H1 (MRS) (28 estudos, 647 esquizofrenia, 608 controle) confirmou diminuição dos níveis de glutamato e aumento dos níveis de glutamina no córtex frontal medial [90]. O papel contributivo da inflamação para a hipoglutamatergia não está comprovado. A síntese elevada de KYNA em cérebros de esquizofrenia [21,52], normalmente uma função da resposta Th2 (Figura 1), pode inibir a subunidade NR1 de NMDAR e alfa 7 nicotínico receptor de acetilcolina (? 7nAchR) [195], levando à diminuição da função NMDAR e redução da liberação de glutamato mediada por? 7nAchR [195].
Na DBP e TOC, os dados sugerem hiperglutamatergia cortical do SNC em ambos os transtornos (Tabela 2) [78,84,88,131]. A contribuição da inflamação (DBP e TOC) e auto-anticorpos (TOC) [7,77,84,88,130] para níveis aumentados de glutamato do SNC requer mais investigação.
O papel de S100B
S100B é uma proteína de ligação ao cálcio de 10 kDa produzida pela astroglia, oligodendroglia e células ependimárias do plexo coróide [196]. Ele medeia seus efeitos nos neurônios circundantes e na glia por meio do receptor para o produto final da glicação avançada [196]. Os níveis extracelulares nanomolares de S100B fornecem efeitos neurotróficos benéficos, limitam a lesão neuronal relacionada ao estresse, inibem o TNF-? liberar e aumentar a recaptação do glutamato astroglial [196]. Concentrações micromolares de S100B, produzidas predominantemente por astroglia e linfócitos ativados [196,197], têm efeitos prejudiciais transduzidos por receptor para produto final de glicação avançada que inclui apoptose neuronal, produção de COX-2 / PGE-2, IL-1? e espécies de óxido nítrico induzíveis e a regulação positiva de TNF-? secreção [21,196,198].
Os níveis séricos e, em particular, de LCR e de tecido cerebral S100B são indicadores da ativação glial (predominantemente astroglial) [199]. Em MDD e psicose, os níveis séricos de S100B correlacionam-se positivamente com a gravidade da suicídio, independentemente do diagnóstico psiquiátrico [200]. A análise post-mortem de S100B mostrou níveis diminuídos no córtex pré-frontal dorso-lateral de MDD e DBP, e níveis aumentados no córtex parietal da DBP [196].
A meta-análise (transtorno de humor 193, controles saudáveis 132) confirmou níveis elevados de soro e CSF S100B em distúrbios de humor, particularmente durante episódios depressivos agudos e mania [201].
Na esquizofrenia, os níveis de cérebro, LCR e S100B sérico estão elevados [199,202]. Meta-análise (12 estudos, 380 esquizofrenia, 358 controles saudáveis) confirmou níveis séricos elevados de S100B na esquizofrenia [203]. Em cérebros post-mortem de indivíduos com esquizofrenia, as astróglias imunorreativas S100B são encontradas em áreas implicadas na esquizofrenia, incluindo córtex cingulado anterior, córtex pré-frontal dorsolateral, córtex orbitofrontal e hipocampo [154]. Níveis elevados de S100B correlacionam-se com psicose paranóide [154] e negativista [204], cognição prejudicada, resposta terapêutica pobre e duração da doença [202]. Polimorfismos genéticos em S100B [32] e receptor para genes de produto final de glicação avançada em coortes de esquizofrenia (Tabela 2) [32,33,205] sugerem que essas anormalidades são provavelmente primárias / patogênicas em vez de secundárias / biomarcadores. De fato, a diminuição nos níveis séricos de S100B após o tratamento com antidepressivos [201] e antipsicóticos [196] sugere alguma relevância clínica de S100B para a fisiopatologia de transtornos psiquiátricos.
Neuroinflamação e aumento do estresse oxidativo
O estresse oxidativo é uma condição em que um excesso de oxidantes danifica ou modifica macromoléculas biológicas, como lipídios, proteínas e DNA [206-209]. Este excesso resulta da produção aumentada de oxidante, diminuição da eliminação de oxidantes, defesas antioxidantes defeituosas ou alguma combinação destes [206-209]. O cérebro é particularmente vulnerável ao estresse oxidativo devido a: 1) quantidades elevadas de ácidos graxos poliinsaturados peroxidizáveis; 2) teor relativamente elevado de minerais traços que induzem peroxidação lipídica e radicais de oxigênio (por exemplo, ferro, cobre); 3) alta utilização de oxigênio; e 3) mecanismos de antioxidação limitados [206,207].
O excesso de estresse oxidativo pode ocorrer em MDD [206], BPD [206,207], esquizofrenia [207,209] e OCD [206,208]. Os marcadores periféricos de distúrbios oxidativos incluem o aumento dos produtos de peroxidação lipídica (por exemplo, malondialdeído e 4-hidroxi-2-não-real), metabolitos aumentados de óxido nítrico (NO), diminuição dos antioxidantes (por exemplo, glutationa) e níveis alterados de enzimas antioxidantes [206,207].
Na MDD, o aumento da produção de aniões radiais de superóxido correlaciona-se com o aumento da apoptose de neutrófilos mediada por oxidação [206]. Os níveis séricos de enzimas antioxidantes (por exemplo, superóxido dismutase-1) são elevados durante episódios depressivos agudos e se normalizam após o tratamento seletivo de inibidores da recaptação da serotonina (SSRIs) [206]. Isso sugere que, na MDD, os níveis séricos de enzimas antioxidantes são um marcador de estado, o que pode refletir um mecanismo compensatório que contraria aumentos agudos no estresse oxidativo. [206]. Na esquizofrenia em contraste, os níveis de superóxido dismutase-1 solúveis em LCR são substancialmente diminuídos nos pacientes com esquizofrenia com início precoce em relação aos pacientes esquizofrênicos crônicos e controles saudáveis. Isso sugere que os níveis reduzidos de enzimas antioxidantes cerebrais podem contribuir para o dano oxidativo na esquizofrenia aguda [210], embora sejam necessários estudos maiores para confirmar essa descoberta.
Vários estudos experimentais e humanos adicionais examinaram com mais detalhes os mecanismos subjacentes à fisiopatologia do aumento do estresse oxidativo em transtornos psiquiátricos [206-262]. Nos modelos animais de depressão, os níveis cerebrais de glutation são reduzidos enquanto a peroxidação lipídica e os níveis de NO aumentam [206,262].
Estudos pós-morte mostram níveis cerebrais reduzidos de glutationa total em MDD, BPD [206] e indivíduos esquizofrênicos [206,207]. Os fibroblastos cultivados a partir de pacientes com MDD apresentam aumento do estresse oxidativo independente dos níveis de glutationa [262], argumentando contra um papel primário da depleção de glutationa como o principal mecanismo de estresse oxidativo na depressão.
A ativação microglial pode aumentar o estresse oxidativo por meio de sua produção de citocinas pró-inflamatórias e NO [206-209]. Citocinas pró-inflamatórias e altos níveis de NO podem promover a formação de espécies reativas de oxigênio (ROS), que por sua vez acelera a peroxidação lipídica, danificando os fosfolipídios da membrana e seus receptores de neurotransmissores monoamínicos ligados à membrana e esgotando os antioxidantes endógenos. Produtos de ROS aumentados podem aumentar a ativação microglial e aumentar a produção pró-inflamatória por meio da estimulação de NF-? B [208], que por sua vez perpetua a lesão oxidativa [208], criando o potencial para um ciclo de feedback positivo patológico em alguns transtornos psiquiátricos [206-209]. Embora a neuroinflamação possa aumentar os níveis de glutamato no cérebro [85,86], o papel da hiperatividade glutamatérgica como causa do estresse oxidativo permanece sem fundamento [207].
A disfunção mitocondrial pode contribuir para o aumento do estresse oxidativo em TDM, DBP e esquizofrenia [206]. Estudos pós-morte nesses distúrbios revelam anormalidades no DNA mitocondrial, consistentes com a alta prevalência de distúrbios psiquiátricos em distúrbios mitocondriais primários [206]. Estudos in vitro em animais mostram que citocinas pró-inflamatórias, como TNF-?, Podem reduzir a densidade mitocondrial e prejudicar o metabolismo oxidativo mitocondrial [211,212], levando ao aumento da produção de ROS [206,213]. Esses achados experimentais podem implicar ligações mecanicistas entre neuroinflamação, disfunção mitocondrial e estresse oxidativo [206,213], merecendo uma investigação mais aprofundada dessas vias patogênicas de intersecção em transtornos psiquiátricos humanos.
A vulnerabilidade do tecido neural ao dano oxidativo varia entre diferentes distúrbios psiquiátricos baseados nas vias neuroanatômicas, neuroquímicas e moleculares envolvidas no distúrbio específico [207]. Os efeitos do tratamento também podem ser críticos, pois evidências preliminares sugerem que antipsicóticos, ISRS e estabilizadores de humor possuem propriedades antioxidantes [206,207,262]. O papel terapêutico dos antioxidantes adjuvantes (por exemplo, vitaminas C e E) em distúrbios psiquiátricos continua a ser fundamentado por ensaios clínicos randomizados de alta potência. A N-acetilcisteína mostra os resultados mais promissores até à data, com vários ensaios randomizados controlados com placebo demonstrando sua eficácia em MDD, DBP e esquizofrenia [207].
Disfunção da barreira hemato-encefálica
O BBB assegura o status de privilégio imunológico do cérebro ao restringir a entrada de mediadores inflamatórios periféricos, incluindo citocinas e anticorpos que podem prejudicar a neurotransmissão [214,215]. A hipótese de quebra da BBB e seu papel em alguns pacientes psiquiátricos [60,214,216,217] é consistente com o aumento da prevalência de comorbidade psiquiátrica em doenças associadas à sua disfunção, incluindo LES [97], acidente vascular cerebral [11], epilepsia [218] e encefalites autoimunes (Tabela 1). Uma elevada taxa de CSF: albumina sérica em pacientes com TDM e esquizofrenia sugere aumento da permeabilidade BBB [214].
Em um estudo (indivíduos 63 psiquiátricos, controles 4,100), alterações do LCR indicativas de dano BBB foram detectadas em 41% de indivíduos psiquiátricos (14 MDD e BPD, esquizofrenia 14), incluindo a síntese intratecal de IgG, IgM e / ou IgA, pleocitose leve de CSF (células 5 a 8 por mm3) e a presença de até quatro bandas oligográficas de IgG [216]. Um estudo ultra-estrutural pós-mortem na esquizofrenia revelou anormalidades ultra-cutâneas BBB nos córtices pré-frontais e visuais, que incluíram degeneração vacuolar de células endoteliais, processos de final de pé astroglial e espessamento e irregularidade da lâmina basal [60]. No entanto, neste estudo, os autores não comentaram sobre o potencial contributo das mudanças pós-morte nas suas descobertas. Outro estudo que investigou transcriptômica de células endoteliais BBB em cérebro esquizofrênico identificou diferenças significativas entre os genes que influenciam a função imunológica, que não foram detectados nos controles [217].
A disfunção endotelial mediada pela oxidação pode contribuir para a fisiopatologia da disfunção BBB em transtornos psiquiátricos. A evidência indireta de estudos clínicos e experimentais na depressão [219] e, em menor grau, na esquizofrenia [220] sugere que o aumento da oxidação pode contribuir para a disfunção endotelial. A disfunção endotelial pode representar um mecanismo compartilhado que representa a associação conhecida entre depressão e doença cardiovascular [219,221], que pode estar relacionada à diminuição dos níveis de vasodilatador NO [221-223]. Estudos experimentais sugerem que os níveis reduzidos de NO endotelial estão ligados mecanicamente ao desacoplamento da óxido nítrico sintase endotelial (eNOS) da sua tetrahidrobiopterina essencial (BH4), deslocando o substrato da L-arginina para o oxigênio [224-226]. O eNOS desacoplado promove a síntese de ROS (por exemplo, superóxido) e espécies de nitrogênio reativo (RNS) (por exemplo, peroxinitrito, produto da interação de superóxido com NO) [227] em vez de NO, levando a disfunção endotelial mediada por oxidação [ 224-226].
Os dados de animais mostraram que os ISRS poderiam restaurar os níveis de NO endoteliais deficientes [219], sugerindo que mecanismos anti-oxidativos podem contribuir para seus efeitos antidepressivos. Em seres humanos, a L-metilfolato pode potencializar os efeitos anti-depressivos dos ISRS [228], de forma voluntária, aumentando os níveis de BH4, que é um cofator essencial para o anti-oxidação mediada por eNOS [229], bem como para a taxa - enzimas limitantes da síntese de monoamina (isto é, serotonina, norepinefrina, dopamina) [228].
Tomados em conjunto, ambos os trabalhos recentes enfatizando o papel do estresse oxidativo induzido por eNOS desacoplado na patogênese de doenças vasculares [230,231] e oEstudos epidemiológicos que estabelecem a depressão como fator de risco independente para patologias vasculares, como acidentes vasculares cerebrais e doença cardíaca [219,221], contribuem ainda mais para a relevância clínica do dano oxidativo endotélio mediado por eNOS desacoplada na depressão. Apesar da abundante evidência de anormalidades de citoquinas em doenças psiquiátricas humanas e os dados experimentais mostrando que as citocinas pró-inflamatórias podem reduzir a expressão de eNOS [212] e aumentar a permeabilidade BBB [215], a evidência humana que relaciona diretamente citocinas pró-inflamatórias excessivas com a disfunção eNOS e / ou deficiência BBB é em falta.
Imagem e tratamento de inflamação em doenças psiquiátricas
Imaging Neuroinflammation In Situ
Clinicamente, a imagem de neuroinflamação pode revelar-se crucial para a identificação do subgrupo de pacientes psiquiátricos com neuroinflamação que provavelmente responderão favoravelmente às terapias imunomoduladoras. Além disso, essa imagem pode permitir que os clínicos monitorem a atividade da doença relacionada à neuroinflamação e sua resposta à terapia imune em pacientes psiquiátricos. A inflamação da imagem no cérebro humano tradicionalmente dependia da visualização de MRI ou CT de agentes de contraste intravenosos extravagados, indicando uma quebra localizada do BBB. A ressonância magnética aumentada de gadolinium ocasionalmente demonstra tal desagregação nas regiões límbicas associadas ao processamento emocional em pacientes com distúrbios psiquiátricos atribuíveis a paraneoplásicos ou outras encefalites [107,109,113]. No nosso conhecimento, no entanto, o aprimoramento anormal nunca foi demonstrado em qualquer transtorno psiquiátrico clássico [21,214,232], apesar das anormalidades funcionais [214,216] e ultraestruturais BBB [60].
Se a neuroinflamação mais sutil pode ou não ser visualizada in vivo em distúrbios psiquiátricos clássicos permanece desconhecida. Uma técnica promissora é a tomografia por emissão de pósitrons (PET) usando radiotracers, como C11-PK11195, que se liga à proteína translocadora, anteriormente conhecida como receptor periférico de benzodiazepina, expressa por microglia ativada [233,234].
Utilizando este método, os pacientes com esquizofrenia demonstraram maior ativação microglial ao longo do córtex [235] e no hipocampo durante psicose aguda [236]. Um estudo (14 esquizofrenia, controles 14) não encontrou diferença significativa entre a ligação [11C] DAA1106 na esquizofrenia versus controles, mas uma correlação direta entre a ligação [11C] DAA1106 e a gravidade dos sintomas positivos e a duração da doença na esquizofrenia [236].
Os investigadores de nossa instituição utilizaram PET C11-PK11195 para demonstrar inflamação bi-hipocampal em um paciente com disfunção neuropsiquiátrica, incluindo TDM psicótico, epilepsia e amnésia anterógrada, associada a anticorpos anti-GAD [237]. No entanto, PK11195 PET tem baixas propriedades de sinal-ruído e requer um ciclotron no local.
Consequentemente, a pesquisa está sendo dedicada ao desenvolvimento de ligandos de proteínas translocadores melhorados para PET e SPECT. Os futuros estudos de tecidos cerebrais pós-mortem de alta potência que utilizam a quantificação de proteínas visando a elucidação das vias metabólicas e inflamatórias, citoquinas do SNC e seus receptores de ligação, em transtornos psiquiátricos são necessários para avançar a nossa compreensão da fisiopatologia auto-imune.
Papel das drogas antiinflamatórias em transtornos psiquiátricos
Vários estudos sobre humanos e animais sugerem que certos medicamentos anti-inflamatórios podem desempenhar um importante papel adjuvante no tratamento de transtornos psiquiátricos (Tabela 3). Os fármacos comuns são inibidores de ciclooxigenase (Tabela 3) [238-245], minociclina (Tabela 3) [240-245], ácidos graxos ômega-3 [246,247] e neuroesteróides [248].
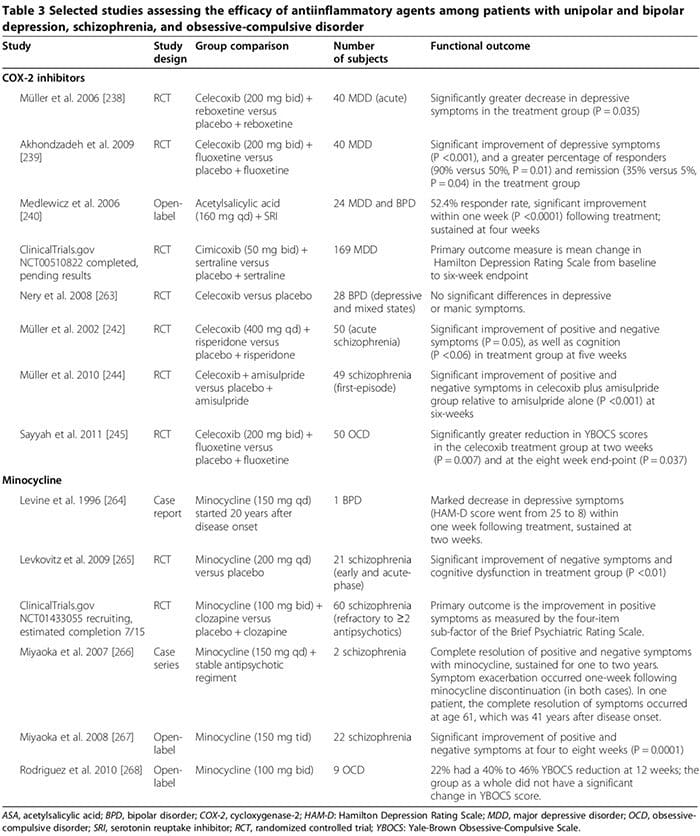 Vários estudos em humanos mostraram que os inibidores de COX-2 poderiam melhorar os sintomas psiquiátricos de MDD, DBP, esquizofrenia e TOC (Tabela 3) [248]. Em contrapartida, o tratamento adjunto com inibidores não selectivos da COX (isto é, antiinflamatórios não esteróides (AINE)) pode reduzir a eficácia dos ISRS [249,250]; dois grandes ensaios relataram que a exposição a AINEs (mas não a inibidores seletivos de COX-2 ou salicilatos) foi associada a um agravamento significativo da depressão entre um subconjunto de participantes do estudo [249,250].
Vários estudos em humanos mostraram que os inibidores de COX-2 poderiam melhorar os sintomas psiquiátricos de MDD, DBP, esquizofrenia e TOC (Tabela 3) [248]. Em contrapartida, o tratamento adjunto com inibidores não selectivos da COX (isto é, antiinflamatórios não esteróides (AINE)) pode reduzir a eficácia dos ISRS [249,250]; dois grandes ensaios relataram que a exposição a AINEs (mas não a inibidores seletivos de COX-2 ou salicilatos) foi associada a um agravamento significativo da depressão entre um subconjunto de participantes do estudo [249,250].
No primeiro teste, envolvendo pacientes deprimidos 1,258 tratados com citalopram por semanas 12, a taxa de remissão foi significativamente menor entre aqueles que haviam tomado NSAIDs pelo menos uma vez em relação àqueles que não tinham (45% versus 55%, OU 0.64, P = 0.0002) [249]. O outro teste, envolvendo indivíduos 1,545 MDD, mostrou que a taxa de depressão resistente ao tratamento foi significativamente maior entre aqueles que tomaram AINEs (OR 1.55, 95% CI 1.21 para 2.00) [231]. O agravamento da depressão nos grupos NSAID pode não estar ligada mecanicamente à terapia com AINEs, mas, em vez disso, relaciona-se a condições médicas crônicas coexistentes [10,12-18] que exigem AINEs de longo prazo e que se sabe que estão independentemente associados com risco aumentado de depressão resistente ao tratamento [249,251]. Estudos futuros que investigam o impacto dos AINEs na depressão e na resposta a antidepressivos em seres humanos são necessários.
Em outros estudos experimentais utilizando paradigmas de estresse agudo para induzir um estado semelhante à depressão em camundongos, o citalopram aumentou TNF- ?, IFN-? E p11 (fator molecular ligado ao comportamento depressivo em animais) no córtex frontal, enquanto o AINE ibuprofeno diminuiu essas moléculas; Os AINEs também atenuaram os efeitos antidepressivos dos SSRIs, mas não de outros antidepressivos [249]. Esses achados sugerem que as citocinas pró-inflamatórias podem, paradoxalmente, exercer efeitos antidepressivos, apesar das evidências esmagadoras de estudos em humanos ao contrário (conforme revisado acima), que podem ser atenuados por AINEs [249]. Pelo menos duas considerações podem ser responsáveis por esse aparente paradoxo: 1) sob algumas condições experimentais, as citocinas pró-inflamatórias foram associadas a um papel neuroprotetor, [251; (para exemplo, IFN-? em níveis baixos pode induzir microglia neuroprotetora (Figura 2) [163,166,251]); e 2) se essas respostas observadas no contexto de um paradigma de estresse agudo em um modelo animal são aplicáveis ao TDM endógeno em humanos permanece obscuro [251].
Os efeitos terapêuticos dos inibidores de COX-2 em transtornos psiquiátricos podem envolver a modulação da biossíntese de prostaglandinas derivadas de COX-2, incluindo PGE2 pró-inflamatória e 15-desoxi-? 12,14-PGJ2 (15d-PGJ2) [252,253]. Os inibidores da COX-2 podem reduzir a inflamação mediada por PGE2, o que pode contribuir para a fisiopatologia dos transtornos psiquiátricos [252,253]. Eles também podem alterar os níveis de 15d-PGJ2 e a atividade de seu receptor nuclear do receptor nuclear ativado por proliferador de peroxissoma (PPAR-?) [252,253].
Vários estudos sugerem que 15d-PGJ2 e seu receptor nuclear PPAR-? podem servir como marcadores biológicos para esquizofrenia [253]. Em pacientes esquizofrênicos, os níveis séricos de PGE2 estão aumentados, enquanto os níveis séricos de 15d-PGJ2 estão diminuídos, assim como a expressão de seu receptor nuclear PPAR-? em PBMC [252]. Embora os inibidores da COX-2 possam limitar os efeitos antiinflamatórios potencialmente benéficos da COX-2 dependente 15d-PGJ2 / PPAR-? pathway , eles podem reduzir vantajosamente seus efeitos prejudiciais, incluindo 1) o risco aumentado de infarto do miocárdio e certas infecções (por exemplo, citomegalovírus e Toxoplasma gondii) em pacientes esquizofrênicos [254] e 2) seus efeitos pró-apoptóticos observados em tecido de câncer humano e animal [255]. Outros mecanismos potenciais dos efeitos terapêuticos dos inibidores da COX-2 podem envolver sua capacidade de reduzir os níveis de citocinas pró-inflamatórias [163], limitar a excitotoxicidade do ácido quinolínico (como no MDD) e diminuir os níveis de KYNA (como na esquizofrenia) [128].
A minociclina pode ser eficaz em transtornos psiquiátricos (Tabela 3) [248]. Dados in vitro sugerem que a minociclina inibe MAP, secreção de citocinas, expressão de COX-2 / PGE-2, e óxido nítrico sintase induzível [256]. A minociclina também pode neutralizar a neurotransmissão glutamatérgica e dopaminérgica desregulada [256].
A eficácia do ácido graxo ômega-3 em transtornos psiquiátricos não é clara [248]. Em uma meta-análise de 2011 de 15 ensaios clínicos randomizados (916 MDD), os suplementos de ômega-3 contendo ácido eicosapentaenóico? 60% (faixa de dose de 200 a 2,200 mg / d em excesso da dose de ácido docosahexaenóico) diminuíram significativamente os sintomas depressivos como um terapia adjuvante aos SRIs (P <0.001) [246]. Uma meta-análise subsequente, entretanto, concluiu que não há benefício significativo dos ácidos graxos ômega-3 na depressão e que a eficácia suposta é meramente um resultado do viés de publicação [247]. Uma meta-análise de 2012 de 5 ensaios clínicos randomizados, incluindo 291 participantes de BPD, descobriu que os sintomas depressivos, mas não maníacos, melhoraram significativamente entre aqueles randomizados para ácidos graxos ômega-3 em relação aos que tomaram placebo (Hedges g 0.34, P = 0.025) [257]. Em um ensaio clínico randomizado com esquizofrênicos acompanhados por até 12 meses, os escores de sintomas positivos e negativos diminuíram significativamente entre os 66 participantes randomizados para ômega-3 de cadeia longa (1.2 g / dia por 12 semanas; P = 0.02 e 0.01, respectivamente) [258]; the os autores concluíram que o aumento de omega-3 durante o início da esquizofrenia também pode prevenir recaídas e progressão da doença [258].
Uma meta-análise 2012 de sete ensaios controlados aleatoriamente que avaliam o aumento de omega-3 em pacientes esquizofrênicos 168 não encontrou nenhum benefício do tratamento [259]. Os autores desta meta-análise declararam especificamente que nenhuma conclusão poderia ser elaborada em relação à prevenção de recaídas ou aos pontos finais da progressão da doença [259]. Os dados experimentais sugerem que o ácido eicosapentaenóico e o ácido docosa-hexaenóico medeiam seus efeitos antiinflamatórios promovendo a síntese de resolvinas e proteinas, o que pode inibir a infiltração de leucócitos e reduzir a produção de citoquinas [248].
Os neuroesteróides, incluindo a pregnenolona e o seu metabolito descendente de alopregnanolona, podem ter um papel benéfico em alguns distúrbios psiquiátricos [248,260]. Na MDD, vários estudos descobriram diminuição dos níveis de plasma / CSF de alopregnanolona correlacionados com a gravidade dos sintomas, que se normalizaram após um tratamento bem-sucedido com certos antidepressivos (por exemplo, SSRIs) e terapia eletroconvulsiva [261]. Na esquizofrenia, os níveis cerebrais de pregnenolona podem ser alterados [248] e os níveis séricos de alopregnanolona podem aumentar após alguns medicamentos antipsicóticos (por exemplo, clozapina e olanzapina) [260]. Em três ensaios controlados aleatoriamente (esquizofrenia 100 (agrupada), duração do tratamento, aproximadamente nove semanas) sintomas positivos, negativos e cognitivos, bem como efeitos colaterais extrapiramidais dos antipsicíticos foram significativamente melhorados em um ou mais ensaios entre aqueles randomizados para pregnenolona em relação aos que receberam placebo [248]. Em um ensaio, a melhoria foi sustentada com o tratamento a longo prazo com pregnenolona [248]. A pregnenolona pode regular a cognição e o comportamento potenciando a função dos receptores NMDA e GABAA [248]. Além disso, a alopregnanolona pode exercer efeitos neuroprotetores e antiinflamatórios [248]. Mais estudos de RCT são necessários para confirmar o papel benéfico dos esteróides neuroactivos em distúrbios psiquiátricos de início precoce em seres humanos.
Estamos aguardando os resultados de vários ensaios clínicos em andamento que investigam os efeitos terapêuticos de outros agentes antiinflamatórios, incluindo salicilato, um inibidor de NF-? B (NCT01182727); ácido acetilsalicílico (NCT01320982); pravastatina (NCT1082588); e dextrometorfano, um antagonista NMDAR não competitivo que pode limitar a lesão neuronal dopaminérgica induzida por inflamação (NCT01189006).
Estratégias de tratamento futuro
Embora as terapias imunológicas atuais (por exemplo, IVIG, plasmaférese, corticosteroides e agentes imunossupressores) sejam frequentemente eficazes para o tratamento de encefalites autoimunes em que a inflamação é aguda, intensa e predominantemente de origem adaptativa, sua eficácia em distúrbios psiquiátricos clássicos em que a inflamação é crônica, muito mais suave e predominantemente de origem inata, é limitado [2]. O desenvolvimento de novas terapêuticas deve visar a reversão da perda glial [46,138], a redução negativa da MAP prejudicial, ao mesmo tempo que otimiza os padrões T neuroprotetivos endógenos e o MAP benéfico, em vez de induzir indiscriminadamente a inflamação, como ocorre com os agentes imunossupressores atuais. Além disso, o desenvolvimento de potentes antioxidantes co-adjuvantes que reverterão a lesão oxidativa em transtornos psiquiátricos é necessário.
Conclusões
A auto-imunidade pode causar uma série de distúrbios neuropsiquiátricos que podem inicialmente apresentar sintomas psiquiátricos isolados. A inflamação inata / auto-imunidade pode ser relevante para a patogênese dos sintomas psiquiátricos em um subconjunto de pacientes com distúrbios psiquiátricos clássicos. A inflamação inata pode estar ligada mecanicamente às anormalidades monoaminérgicas e glutamatérgicas tradicionais e ao aumento da lesão oxidativa relatada em doenças psiquiátricas.
Souhel Najjar1,5 *, Daniel M Pearlman2,5, Kenneth Alper4, Amanda Najjar3 e Orrin Devinsky1,4,5
Abreviaturas
3-OH-KYN: 3-hidroxi-quinurenina; ? 7nAchR: receptores nicot�icos alfa 7 de acetilcolina; AMPAR: Receptores de ácido amino-3-hidroxi-5-metil-l-4-isoxazolpropiônico; APC: Célula apresentadora de antígeno; BBB: Barreira hematoencefálica;
BH4: Tetrahidrobiopterina; DBP: transtorno bipolar; CI: intervalo de confiança;
CNS: sistema nervoso central; COX-2: Cyclooxegenase-2; LCR: líquido cefalorraquidiano; DSM-IV: Manual de Diagnóstico e Estatística de Distúrbios Mentais 4th Edition; EAATs: transportadores excitadores de aminoácidos; eNOS: óxido nítrico sintase endotelial; GABAB: ácido gamma aminobutírico-beta; GAD: Decarboxilase de ácido glutâmico; GFAP: proteína ácida fibrilar glial; GLX: 1H MRS glutamato detectável, glutamina, ácido gama aminobutírico composto;
IDO: Indoleamina 2,3-dioxigenase; Ig: imunoglobulina; IL: Interleucina; IL-1RA: antagonista do receptor da interleucina 1; IFN- ?: Interferon gama;
KAT: Kynurenine aminotransferase; KMO: Kynurenine 3-monooxygenase; KYN: Kynurenine; KYNA: ácido Kynurenic; LE: encefalite límbica;
LPS: lipopolissacarídeo; MAP: ativação e proliferação microglial;
TDM: transtorno depressivo maior; mGluR: receptor metabotrópico de glutamato; MHC: II Complexo principal de histocompatibilidade classe dois; MRI: Imagem por ressonância magnética; MRS: espectroscopia de ressonância magnética; NF-? B: Fator nuclear kappa B; NMDAR: receptor de N-metil-D-aspartato; NR1: local da glicina;
TOC: transtorno obsessivo-compulsivo; OR: odds ratio; PANDAS: distúrbios auto-imunes neuropsiquiátricos pediátricos associados a infecções estreptocócicas; PBMC: células mononucleares do sangue periférico; PET: tomografia por emissão de positrões; PFC: córtex pré-frontal; PGE-2: Prostaglandina E2; PPAR-
?: Receptor nuclear gama ativado por proliferador de peroxissoma; QA: ácido quinolínico; RNS: espécies reativas de nitrogênio; ROS: espécies reativas de oxigênio;
sIL: Interleucina solúvel; LES: lúpus eritematoso sistêmico; SRI: inibidor da recaptação da serotonina; TNF- ?: Fator de necrose tumoral alfa; T-regs: células T reguladoras CD4 + CD25 + FOXP3 +; TDO: Triptofano-2,3-dioxigenase; Th: T-helper; VGKC: Canal de potássio controlado por voltagem; XAG-: transportador de glutamato aspartato; Xc-: glutamato / cistina astroglial independente de sódio
sistema antiterrorista
Interesses competitivos
Os autores declaram que não têm interesses concorrentes.
Autores Contribuições
SN e DMP realizaram uma extensa revisão da literatura, dados interpretados, prepararam o manuscrito, figuras e tabelas. A KA preparou a seção referente aos mecanismos oxidativos e contribuiu para as revisões do manuscrito. AN e OD criticamente revisaram e melhoraram o design e a qualidade do manuscrito. Todos os autores leram e aprovaram o manuscrito final.
Agradecimentos
Agradecemos aos Drs. Josep Dalmau, MD, PhD, Tracy Butler, MD e David Zazag, MD, PhD, por fornecerem sua experiência em encefalites autoimunes, neuroinflamação e neuropatologia, respectivamente.
Autor Detalhes
1 Departamento de Neurologia, Faculdade de Medicina da Universidade de Nova York, 550 First Avenue, Nova York, NY 10016, EUA. 2Geisel School of Medicine em Dartmouth, Instituto Dartmouth de Políticas de Saúde e Prática Clínica, 30 Lafayette Street, HB 7252, Líbano, NH 03766, EUA. 3 Departamento de Patologia, Divisão de Neuropatologia, Faculdade de Medicina da Universidade de Nova York, 550 First Avenue, Nova York, NY 10016, EUA. 4 Departamento de Psiquiatria, Faculdade de Medicina da Universidade de Nova York, Nova York, NY, EUA. 5 New York University Comprehensive Epilepsy Center, 550 First Avenue, Nova York, NY 10016, EUA.
Blank
Referências:
1. Kayser MS, Dalmau J: O elo emergente entre distúrbios auto-imunes
e doença neuropsiquiátrica. J Neuropsychiatry Clin Neurosci 2011, 23: 90 97.
2. Najjar S, Pearlman D, Zagzag D, Golfinos J, Devinsky O: ácido glutâmico
síndrome de autoanticorpo de descarboxilase apresentando-se como esquizofrenia.
Neurologist 2012, 18: 88 91.
3. Graus F, Saiz A, Dalmau J: Anticorpos e auto-imune neuronal
distúrbios do SNC. J Neurol 2010, 257: 509 517.
4. Lennox BR, Coles AJ, Vincent A: encefalite mediada por anticorpos: a
causa tratável de esquizofrenia. Br J Psychiatry 2012, 200: 92 94.
5. Zandi MS, Irani SR, Lang B, Waters P, Jones PB, McKenna P, Coles AJ, Vincent
A, Lennox BR: Autoanticorpos relevantes para a doença no primeiro episódio
esquizofrenia. J Neurol 2011, 258: 686 688.
6. Bataller L, Kleopa KA, Wu GF, Rossi JE, Rosenfeld MR, Dalmau J:
Encefalite límbica autoimune em pacientes com 39: imunofenotipos e
resultados. J Neurol Neurosurg Psychiatry 2007, 78: 381 385.
7. Dale RC, Heyman I, Giovannoni G, Igreja AW: Incidência de anti-cérebro
anticorpos em crianças com transtorno obsessivo-compulsivo. Br J Psiquiatria
2005, 187: 314 319.
8. Kendler KS: A natureza dappled das causas da doença psiquiátrica: substituindo
a dicotomia orgânico-funcional / hardware-software com empiricamente
pluralismo baseado. Mol Psychiatry 2012, 17: 377 388.
9. Keskin G, Sunter G, Midi I, Tuncer N: Neurosyphilis como causa de cognitivo
declínio e sintomas psiquiátricos na idade mais jovem. J Neuropsychiatry Clin
Neurosci 2011, 23: E41 E42.
10. Leboyer M, Soreca I, Scott J, Frye M, Henry C, Tamouza R, Kupfer DJ: Can
o transtorno bipolar deve ser visto como uma doença inflamatória multi-sistema?
J Affect Disord 2012, 141: 1 10.
11. Hackett ML, Yapa C, Parag V, Anderson CS: Freqüência de depressão após
AVC: uma revisão sistemática de estudos observacionais. Stroke 2005, 36: 1330 1340.
12. Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW: de
inflamação da doença e depressão: quando o sistema imunológico
subjuga o cérebro. Nat Rev Neurosci 2008, 9: 46 56.
13. Laske C, Zank M, Klein R, Stransky E, Batra A, Buchkremer G, Schott K:
Reatividade do autoanticorpo no soro de pacientes com depressão maior,
esquizofrenia e controles saudáveis. Psychiatry Res 2008, 158: 83 86.
14. Eisenberger NI, Berkman ET, Inagaki TK, Rameson LT, Mashal NM, Irwin MR:
Anedonia induzida por inflamação: a endotoxina reduz o estriado ventral
respostas à recompensa. Biol Psychiatry 2010, 68: 748 754.
15. Haroon E, Raison CL, Miller AH: Psychoneuroimmunology atende
neuropsicofarmacologia: implicações translacionais do impacto de
inflamação no comportamento. Neuropsychopharmacology 2012, 37: 137 162.
16. Benros ME, Nielsen PR, Nordentoft M, Eaton WW, Dalton SO, Mortensen PB:
Doenças auto-imunes e infecções graves como fatores de risco para
esquizofrenia: estudo de registro baseado em população 30-year. Am J Psiquiatria
2011, 168: 1303 1310.
17. McNally L, Bhagwagar Z, Hannestad J: inflamação, glutamato e glia
na depressão: uma revisão da literatura. CNS Spectr 2008, 13: 501 510.
18. Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Critchley HD:
A inflamação provoca mudanças de humor através de alterações no subgenual
atividade cingulada e conectividade mesolímbica. Biol Psiquiatria 2009,
66: 407 414.19. Raison CL, Miller AH: A depressão é um distúrbio inflamatório?
Curr Psychiatry Rep 2011, 13: 467 475.
20. Raison CL, Miller AH: o significado evolutivo da depressão em
Defesa do Host de Patógenos (PATHOS-D). Mol Psychiatry 2013, 18: 15 37.
21. Steiner J, Bogerts B, Sarnyai Z, Walter M, Gos T, Bernstein HG, Myint AM:
Colidindo o fosso entre as hipóteses imune e glutamato de
esquizofrenia e depressão maior: papel potencial do NMDA glial
moduladores de receptor e integridade da barreira hematoencefálica prejudicada. Mundo J
Biol Psychiatry 2012, 13: 482 492.
22. Steiner J, Mawrin C, Ziegeler A, Bielau H, Ullrich O, Bernstein HG, Bogerts B:
A distribuição da microglia HLA-DR positiva na esquizofrenia reflete
lateralização cerebral prejudicada. Acta Neuropathol 2006, 112: 305 316.
23. Papakostas GI, Shelton RC, Kinrys G, Henry ME, Bakow BR, Lipkin SH, Pi B,
Thurmond L, Bilello JA: avaliação de um teste múltiplo, baseado em soro
Teste de diagnóstico biológico para transtorno depressivo maior: um piloto e
estudo de replicação. Mol Psychiatry 2013, 18: 332 339.
24. Krishnan R: depressão unipolar em adultos: epidemiologia, patogênese e
neurobiologia. No UpToDate. Editado por Basow DS. Waltham, MA: UpToDate; 2013.
25. Stovall J: Transtorno bipolar em adultos: epidemiologia e diagnóstico. Dentro
Atualizado. Editado por Basow DS. UpToDate: Waltham; 2013.
26. Fischer BA, Buchanan RW: esquizofrenia: epidemiologia e patogênese.
No UpToDate. Editado por Basow DS. Waltham, MA: UpToDate; 2013.
27. Nestadt G, Samuels J, Riddle M, Bienvenu OJ 3rd, Liang KY, LaBuda M,
Walkup J, Grados M, Hoehn-Saric R: estudo familiar de obsessivecompulsive
transtorno. Arch Gen Psychiatry 2000, 57: 358 363.
28. Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D,
Werge T, Pietilainen OP, Mors O, Mortensen PB, Sigurdsson E, Gustafsson O,
Nyegaard M, Tuulio-Henriksson A, Ingason A, Hansen T, Suvisaari J,
Lonnqvist J, Paunio T, B rglum AD, Hartmann A, Fink-Jensen A, Nordentoft
M, Hougaard D, Norgaard-Pedersen B, B ttcher Y, Olesen J, Breuer R, M ller
HJ, Giegling I, et al: Variantes comuns que conferem risco de esquizofrenia.
Nature 2009, 460: 744 747.
29. M ller N, Schwarz MJ: A alteração imunomediada da serotonina e
glutamato: para uma visão integrada da depressão. Mol Psiquiatria
2007, 12: 988 1000.
30. Galecki P, Florkowski A, Bienkiewics M, Szemraj J: polimorfismo funcional
do gene da ciclooxigenase-2 (G-765C) em pacientes depressivos.
Neuropsychobiology 2010, 62: 116 120.
31. Levinson DF: A genética da depressão: uma revisão. Biol Psiquiatria 2006,
60: 84 92.
32. Zhai J, Cheng L, Dong J, Shen Q, Zhang Q, Chen M, Gao L, Chen X, Wang K,
Deng X, Xu Z, Ji F, Liu C, Li J, Dong Q, Chen C: gene S100B
Os polimorfismos prevêem a função espacial pré-frontal tanto na esquizofrenia
pacientes e indivíduos saudáveis. Schizophr Res 2012, 134: 89 94.
33. Zhai J, Zhang Q, Cheng L, Chen M, Wang K, Liu Y, Deng X, Chen X, Shen Q,
Xu Z, Ji F, Liu C, Dong Q, Chen C, Li J: Variantes de risco no gene S100B,
associados a níveis elevados de S100B, também estão associados a
deficiência visuoespacial da esquizofrenia. Behav Brain Res 2011, 217: 363 368.
34. Cappi C, Muniz RK, Sampaio AS, Cordeiro Q, Brentani H, Palacios SA,
Marques AH, Vallada H, Miguel EC, Guilherme L, Hounie AG: Associação
estudo entre polimorfismos funcionais no gene TNF-alfa e
transtorno obsessivo-compulsivo. Arq Neuropsiquiatr 2012, 70: 87 ± 90.
35. Miguel-Hidalgo JJ, Baucom C, Dilley G, Overholser JC, Meltzer HY,
Stockmeier CA, Rajkowska G: proteína acídica fibrilante glial
A imunorreatividade no córtex pré-frontal distingue mais jovens de
adultos mais velhos com transtorno depressivo maior. Biol Psychiatry 2000, 48: 861 873.
36. Altshuler LL, Abulseoud OA, Foland Ross L, Bartzokis G, Chang S, Mintz J,
Hellemann G, Vinters HV: redução de astrocitos de Amígdala em indivíduos com
transtorno depressivo maior, mas não transtorno bipolar. Bipolar Disord 2010,
12: 541 549.
37. Webster MJ, Knable MB, Johnston-Wilson N, Nagata K, Inagaki M, Yolken RH:
Localização imuno-histoquímica de ácido fibrilante glial fosforilado
proteína no córtex pré-frontal e hipocampo de pacientes com
esquizofrenia, transtorno bipolar e depressão. Brain Behav Immun 2001,
15: 388 400.
38. Doyle C, Deakin JFW: Menos astrocitos no córtex frontal na esquizofrenia,
depressão e transtorno bipolar. Esquizofrenia Res 2002, 53: 106.
39. Johnston-Wilson NL, Sims CD, Hofmann JP, Anderson L, Shore AD, Torrey
EF, Yolken RH: alterações específicas da doença no córtex frontal proteínas do cérebro
na esquizofrenia, transtorno bipolar e transtorno depressivo maior, The
Stanley Neuropathology Consortium. Mol Psychiatry 2000, 5: 142 149.
40. Gosselin RD, Gibney S, O'Malley D, Dinan TG, Cryan JF: específico da região
diminuição da imunorreactividade da proteína ácida fibrilante glial no cérebro de
um modelo de rato de depressão. Neuroscience 2009, 159: 915 925.
41. Banasr M, Duman RS: perda glial no córtex pré-frontal é suficiente para
induzir comportamentos semelhantes aos depressivos. Biol Psychiatry 2008, 64: 863 870.
42. Cotter D, Hudson L, Landau S: evidência de patologia orbito-frontal em
transtorno bipolar e depressão maior, mas não na esquizofrenia.
Bipolar Disord 2005, 7: 358 369.
43. Brauch RA, Adnan El-Masri M, Parker J Jr, El-Mallakh RS: número de células gliales
e taxas de células neuronais / gliais em cérebros pós-morte de indivíduos bipolares.
J Affect Disord 2006, 91: 87 90.
44. Cotter DR, Pariante CM, Everall IP: anormalidades das células gliais em grandes
transtornos psiquiátricos: evidências e implicações. Brain Res Bull 2001,
55: 585 595.
45. Cotter D, Mackay D, Landau S, Kerwin R, Everall I: densidade de células gliais reduzidas
e tamanho neuronal no córtex cingulado anterior em depressão maior
transtorno. Arch Gen Psychiatry 2001, 58: 545 553.
46. Bowley MP, Drevets WC, Ong r D, Price JL: Números gliais baixos no
amígdala no transtorno depressivo maior. Biol Psychiatry 2002, 52: 404 412.
47. Toro CT, Hallak JE, Dunham JS, Deakin JF: proteína ácida fibrilar glial e
glutamina sintetase nas sub-regiões do córtex pré-frontal na esquizofrenia
e transtorno de humor. Neurosci Lett 2006, 404: 276 281.
48. Rajkowska G, Miguel-Hidalgo JJ, Makkos Z, Meltzer H, Overholser J,
Stockmeier C: reduções específicas da camada na astroglia reactiva ao GFAP no
córtex pré-frontal dorsolateral na esquizofrenia. Schizophr Res 2002, 57: 127 138.
49. Steffek AE, McCullumsmith RE, Haroutunian V, Meador-Woodruff JH: Cortical
A expressão de proteína ácida fibrilar glial e glutamina sintetase é
diminuiu na esquizofrenia. Schizophr Res 2008, 103: 71 82.
50. Damadzic R, Bigelow LB, Krimer LS, Goldenson DA, Saunders RC, Kleinman
JE, Herman MM: estudo quantitativo imuno-histoquímico de astrocitos em
o córtex entorrinal na esquizofrenia, transtorno bipolar e maior
depressão: ausência de astrocitose significativa. Brain Res Bull 2001, 55: 611 618.
51. Benes FM, McSparren J, Bird ED, SanGiovanni JP, Vincent SL: Déficits em
Pequenos interneurônios em córtex pré-frontal e cingulado de esquizofrênico
e pacientes esquizoafetivos. Arch Gen Psychiatry 1991, 48: 996 1001.
52. M ller N, Schwarz MJ: Sistema imunológico e esquizofrenia. Curr Immunol
Rev 2010, 6: 213 220.
53. Steiner J, Walter M, Gos T, Guillemin GJ, Bernstein HG, Sarnyai Z, Mawrin C,
Brisch R, Bielau H, Meyer Zu Schwabedissen L, Bogerts B, Myint AM: Grave
A depressão está associada ao aumento do ácido quinolínico microglial em
sub-regiões do giro cingulado anterior: evidência de um imunododulador
neurotransmissão glutamatérgica? J Neuroinflamação 2011, 8: 94.
54. Vostrikov VM, Uranova NA, Orlovskaya DD: Déficit de perineuronal
oligodendrócitos no córtex pré-frontal na esquizofrenia e humor
desordens. Schizophr Res 2007, 94: 273 280.
55. Rajkowska G, Miguel-Hidalgo JJ: Gliogênese e patologia glial em
depressão. CNS Neurol Disord Drug Targets 2007, 6: 219 ± 233.
56. Uranova NA, Vostrikov VM, Orlovskaya DD, Rachmanova VI:
Densidade oligodendroglial no córtex pré-frontal em esquizofrenia e
transtornos do humor: um estudo do Consórcio de Neuropatologia Stanley.
Schizophr Res 2004, 67: 269 275.
57. Uranova N: danos e perda de oligodendrócitos são cruciais no
patogênese da esquizofrenia e distúrbios do humor (forma de descoberta
estudos pós-morte). Neuropsicofarmacologia 2004, 29: S33.
58. Uranova NA, Orlovskaya DD, Vostrikov VM, Rachmanova VI: Diminuição
densidade de satélites oligodendrogliais de neurônios piramidais na camada III em
o córtex pré-frontal na esquizofrenia e distúrbios do humor. Schizophr Res
2002, 53: 107.
59. Vostrikov VM, Uranova NA, Rakhmanova VI, Orlovskaia DD: Abaixou
densidade de células oligodendrogliais no córtex pré-frontal na esquizofrenia.
Zh Nevrol Psikhiatr Im SS Korsakova 2004, 104: 47 51.
60. Uranova NA, Zimina IS, Vikhreva OV, Krukov NO, Rachmanova VI, Orlovskaya
DD: danos ultraestruturais dos capilares no neocórtex em
esquizofrenia. World J Biol Psychiatry 2010, 11: 567 578.
61. Hof PR, Haroutunian V, Friedrich VL Jr, Byne W, Buitron C, Perl DP, Davis KL:
Perda e distribuição espacial alterada de oligodendrócitos no superior
giro frontal na esquizofrenia. Biol Psychiatry 2003, 53: 1075 1085.
62. Davis KL, Stewart DG, Friedman JI, Buchsbaum M, Harvey PD, Hof PR,
Buxbaum J, Haroutunian V: alterações da matéria branca na esquizofrenia:
evidência de disfunção relacionada com mielina. Arc Gen Psychiatry 2003,
60: 443 456.63. Flynn SW, Lang DJ, Mackay AL, Goghari V, Vavasour IM, Whittall KP, Smith
GN, Arango V, Mann JJ, Dwork AJ, Falkai P, Honer WG: anormalidades de
mielinização na esquizofrenia detectada in vivo com ressonância magnética e pós-morte
com análise de proteínas oligodendrocitadas. Mol Psychiatry 2003,
8: 811 820.
64. Uranova NA, Vostrikov VM, Vikhreva OV, Zimina IS, Kolomeets NS, Orlovskaya
DD: O papel da patologia oligodendrocitária na esquizofrenia. Int J
Neuropsychopharmacol 2007, 10: 537 ± 545.
65. Byne W, Kidkardnee S, Tatusov A, Yiannoulos G, Buchsbaum MS,
Haroutunian V: redução associada à esquizofrenia de neuronais e
números de oligodendrócitos no núcleo talâmico principal anterior.
Schizophr Res 2006, 85: 245 253.
66. Hamidi M, Drevets WC, Preço JL: redução glial na amígdala em grande
transtorno depressivo é devido a oligodendrócitos. Biol Psiquiatria 2004,
55: 563 569.
67. Bayer TA, Buslei R, Havas L, Falkai P: Evidências de ativação da microglia em
pacientes com doenças psiquiátricas. Neurosci Lett 1999, 271: 126 128.
68. Steiner J, Bielau H, Brisch R, Danos P, Ullrich O, Mawrin C, Bernstein HG,
Bogerts B: Aspectos imunológicos na neurobiologia do suicídio:
elevada densidade microglial na esquizofrenia e depressão é
associado ao suicídio. J Psychiatr Res 2008, 42: 151 157.
69. Rao JS, Harry GJ, Rapoport SI, Kim HW: aumento da excitotoxicidade e
marcadores neuroinflamatórios no córtex frontal pós-mortem de bipolar
pacientes com transtorno. Mol Psychiatry 2010, 15: 384 392.
70. Bernstein HG, Steiner J, Bogerts B: células gliales na esquizofrenia:
significado fisiopatológico e possíveis consequências para a terapia.
Expert Rev Neurother 2009, 9: 1059 ± 1071.
71. Chen SK, Tvrdik P, Peden E, Cho S, Wu S, Spangrude G, Capecchi MR:
Origem hematopoiética de tratamento patológico em camundongos mutantes Hoxb8.
Cell 2010, 141: 775 785.
72. Antony JM: Higiene e crescimento com microglia. Sci Signal 2010, 3: jc8.
73. Wonodi I, Stine OC, Sathyasaikumar KV, Roberts RC, Mitchell BD, Hong LE,
Kajii Y, Thaker GK, Schwarcz R: Kynurenine com Downregulated 3-
expressão do gene da monoxigenase e atividade enzimática na esquizofrenia
e associação genética com endófenotipos de esquizofrenia. Arch Gen
Psychiatry 2011, 68: 665 674.
74. Raison CL, Lowry CA, Rook GA: inflamação, saneamento e
consternação: perda de contato com coevolved, tolerogênico
microorganismos e a fisiopatologia e tratamento de grandes
depressão. Arch Gen Psychiatry 2010, 67: 1211 1224.
75. Drexhage RC, Hoogenboezem TH, Versnel MA, Berghout A, Nolen WA,
Drexhage HA: A ativação de redes de monocitos e células T em pacientes
com transtorno bipolar. Brain Behav Immun 2011, 25: 1206 1213.
76. Steiner J, Jacobs R, Panteli B, Brauner M, Schiltz K, Bahn S, Herberth M,
Westphal S, Gos T, Walter M, Bernstein HG, Myint AM, Bogerts B: aguda
A esquizofrenia é acompanhada por células T reduzidas e células B aumentadas
imunidade. Eur Arch Psychiatry Clin Neurosci 2010, 260: 509 ± 518.
77. Rotge JY, Aouizerate B, Tignol J, Bioulac B, Burbaud P, Guehl D: O
Hipótese imune genética baseada em glutamato em obsessivo-compulsivo
Desordem, Uma abordagem integrativa dos genes aos sintomas.
Neuroscience 2010, 165: 408 417.
78. Y ksel C, Ong r D: Estudos de espectroscopia de ressonância magnética de
anormalidades relacionadas ao glutamato nos distúrbios do humor. Biol Psiquiatria 2010,
68: 785 794.
79. Rao JS, Kellom M, Reese EA, Rapoport SI, Kim HW: glutamato desregulado
e transportadores de dopamina no córtex frontal pós-morte de bipolar
e pacientes esquizofrênicos. J Affect Disord 2012, 136: 63 71.
80. Bauer D, Gupta D, Harotunian V, Meador-Woodruff JH, McCullumsmith RE:
Expressão anormal do transportador e transportador de glutamato
moléculas interagindo no córtex pré-frontal em pacientes idosos com
esquizofrenia. Schizophr Res 2008, 104: 108 120.
81. Matute C, Melone M, Vallejo-Illarramendi A, Conti F: Aumento da expressão
do transportador de glutamato astrocítico GLT-1 no córtex pré-frontal de
esquizofrênicos. 2005, 49: 451 455.
82. Smith RE, Haroutunian V, Davis KL, Meador-Woodruff JH: Expressão de
transcritos excitatórios do transportador de aminoácidos no tálamo de indivíduos
com esquizofrenia. Am J Psychiatry 2001, 158: 1393 1399.
83. McCullumsmith RE, Meador-Woodruff JH: aminoácido excitatório de Striatal
expressão transcrita do transportador na esquizofrenia, transtorno bipolar
e transtorno depressivo maior. Neuropsicofarmacologia 2002,
26: 368 375.
84. Pittenger C, Bloch MH, Williams K: anormalidades de glutamato em obsessivo
transtorno compulsivo: neurobiologia, fisiopatologia e tratamento.
Pharmacol Ther 2011, 132: 314 332.
85. Hashimoto K: papel emergente do glutamato na fisiopatologia de
transtorno depressivo maior. Brain Res Rev 2009, 61: 105 123.
86. Hashimoto K, Sawa A, Iyo M: Aumento dos níveis de glutamato nos cérebros de
pacientes com transtornos de humor. Biol Psychiatry 2007, 62: 1310 1316.
87. Burbaeva G, Boksha IS, Turishcheva MS, Vorobyeva EA, Savushkina OK,
Tereshkina EB: Glutamina sintetase e glutamato desidrogenase em
o córtex pré-frontal de pacientes com esquizofrenia. Prog
Neuropsychopharmacol Biol Psychiatry 2003, 27: 675 680.
88. Bhattacharyya S, Khanna S, Chakrabarty K, Mahadevan A, Christopher R,
Shankar SK: autoanticorpos anti-cérebro e excitatório alterado
neurotransmissores em transtorno obsessivo-compulsivo.
Neuropsychopharmacology 2009, 34: 2489 2496.
89. Sanacora G, Gueorguieva R, Epperson CN, Wu YT, Appel M, Rothman DL,
Krystal JH, Mason GF: alterações específicas do subtipo de gammaaminobutyric
ácido e glutamato em pacientes com depressão maior.
Arch Gen Psychiatry 2004, 61: 705 713.
90. Marsman A, van den Heuvel MP, Klomp DW, Kahn RS, Luijten PR, Hulshoff
Pol HE: Glutamato na esquizofrenia: uma revisão focalizada e metanálise
de estudos de 1H-MRS. Schizophr Bull 2013, 39: 120 129.
91. Liu Y, Ho RC, Mak A: Interleucina (IL) -6, fator de necrose tumoral alfa
(TNF-alfa) e os receptores solubles de interleucina-2 (sIL-2R) estão elevados em
pacientes com transtorno depressivo maior: uma meta-análise e metarrevisão.
J Affect Disord 2012, 139: 230 239.
92. Brietzke E, Stabellini R, Grassis-Oliveira R, Lafer B: citocinas em bipolar
desordem: descobertas recentes, efeitos deletérios, mas promessa para o futuro
terapêutica. CNS Spectr 2011. www.cnsspectrums.com/aspx/
articledetail.aspx? articleid = 3596.
93. Denys D, Fluitman S, Kavelaars A, Heijnen C, Westenberg H: Diminuição
Actividade TNF-alfa e NK no transtorno obsessivo-compulsivo.
Psychoneuroendocrinology 2004, 29: 945 952.
94. Brambilla F, Perna G, Bellodi L, Arancio C, Bertani A, Perini G, Carraro C, Gava
F: concentrações plasmáticas de interleucina-1 beta e de necrose tumoral em
transtornos obsessivo-compulsivos. Biol Psychiatry 1997, 42: 976 981.
95. Fluitman S, Denys D, Vulink N, Schutters S, Heijnen C, Westenberg H:
Produção de citoquinas induzida por lipopolisacarídeos em obsessivecompulsiva
desordem e transtorno de ansiedade social generalizada. Psiquiatria
Res 2010, 178: 313 316.
96. Janelidze S, Mattei D, Westrin A, Traskman-Bendz L, Brundin L: Citocinas
níveis no sangue podem distinguir tentadores de suicídios deprimidos
pacientes. Brain Behav Immun 2011, 25: 335 339.
97. Postal M, Costallat LT, Appenzeller S: manifestações neuropsiquiátricas em
Lúpus eritematoso sistêmico: epidemiologia, fisiopatologia e
gestão. CNS Drugs 2011, 25: 721 736.
98. Kozora E, Hanly JG, Lapteva L, Filley CM: disfunção cognitiva em
Lúpus eritematoso sistêmico: passado, presente e futuro.
Arthritis Rheum 2008, 58: 3286 3298.
99. Lancaster E, Martinez-Hernandez E, Dalmau J: Encefalite e anticorpos para
proteínas da superfície celular sináptica e neuronal. Neurology 2011, 77: 179 189.
100. Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon
R: Experiência clínica e pesquisas laboratoriais em pacientes com antiNMDAR
encefalite. Lancet Neurol 2011, 10: 63 74.
101. Lai M, Huijbers MG, Lancaster E, Graus F, Bataller L, Balice-Gordon R, Cowell
JK, Dalmau J: Investigação de LGI1 como antígeno na encefalite límbica
anteriormente atribuído aos canais de potássio: uma série de casos. Lancet Neurol
2010, 9: 776 785.
102. Lancaster E, Huijbers MG, Bar V, Boronat A, Wong A, Martinez-Hernandez E,
Wilson C, Jacobs D, Lai M, Walker RW, Graus F, Bataller L, Illa I, Markx S, Strauss
KA, Peles E, Scherer SS, Dalmau J: investigações de caspr2, um autoantigênio de
encefalite e neuromiotonia. Ann Neurol 2011, 69: 303 311.
103. Lancaster E, Lai M, Peng X, Hughes E, Constantinescu R, Raizer J, Friedman
D, Skeen MB, Grisold W, Kimura A, Ohta K, Iizuka T, Guzman M, Graus F,
Moss SJ, Balice-Gordon R, Dalmau J: anticorpos para o receptor GABA (B) em
encefalite límbica com convulsões: série de casos e caracterização da
antígeno. Lancet Neurol 2010, 9: 67 76.
104. Lancaster E, Martinez-Hernandez E, Titulaer MJ, Boulos M, Weaver S, Antoine
JC, Liebers E, Kornblum C, Bien CG, Honnorat J, Wong S, Xu J, contratado A,
Balice-Gordon R, Dalmau J: anticorpos para o glutamato metabotrópico
receptor 5 na síndrome de Ophelia. Neurology 2011, 77: 1698 1701.105. Ances BM, Vitaliani R, Taylor RA, Liebeskind DS, Voloschin A, Houghton DJ,
Galetta SL, Dichter M, Alavi A, Rosenfeld MR, Dalmau J: Tratamento responsivo
encefalite límbica identificada por anticorpos neuropil: ressonância magnética e
PET correlacionados. Brain 2005, 128: 1764 1777.
106. Tofaris GK, Irani SR, Cheeran BJ, Baker IW, Cader ZM, Vincent A:
Coréia responsiva à imunoterapia como característica de apresentação da LGI1-
encefalite por anticorpos. Neurology 2012, 79: 195 196.
107. Najjar S, Pearlman D, Najjar A, Ghansian V, Zagzag D, Devinsky O:
Encefalite autoimune extralimática associada com ácido glutâmico
Anticorpos de descarboxilase: uma entidade subdiagnosticada? Epilepsy Behav
2011, 21: 306 313.
108. Titulaer MJ, McCracken L, Gabilondo I, Armangue T, Glaser C, Iizuka T, Honig
LS, Benseler SM, Kawachi I, Martinez-Hernandez E, Aguilar E, Gresa-Arribas N,
Ryan-Florance N, Torrents A, Saiz A, Rosenfeld MR, Balice-Gordon R, Graus F,
Dalmau J: Tratamento e fatores prognósticos para o desfecho a longo prazo em
Pacientes com encefalite anti-receptor NMDA: uma coorte observacional
estudar. Lancet Neurol 2013, 12: 157 165.
109. Dalmau J, Gleichman AJ, Hughes EG, Rossi JE, Peng X, Lai M, Dessain SK,
Rosenfeld MR, Balice-Gordon R, Lynch DR: receptor anti-NMDA
encefalite: série de casos e análise dos efeitos de anticorpos.
Lancet Neurol 2008, 7: 1091 1098.
110. Graus F, Boronat A, Xifro X, Boix M, Svigelj V, Garcia A, Palomino A, Sabater
L, Alberch J, Saiz A: o perfil clínico em expansão do receptor anti-AMPA
encefalite. Neurology 2010, 74: 857 859.
111. Lai M, Hughes EG, Peng X, Zhou L, Gleichman AJ, Shu H, Mata S, Kremens
D, Vitaliani R, Geschwind MD, Bataller L, Kalb RG, Davis R, Graus F, Lynch DR,
Balice-Gordon R, Dalmau J: anticorpos do receptor AMPA em limbic
a encefalite altera a localização do receptor sináptico. Ann Neurol 2009, 65: 424 434.
112. Najjar S, Pearlman D, Devinsky O, Najjar A, Nadkarni S, Butler T, Zagzag D:
Encefalite auto-imune neuropsiquiátrica com complexo VGKC negativo,
NMDAR e autoanticorpos GAD: um relato de caso e revisão de literatura,
próximo. Cogn Behav Neurol. na imprensa.
113. Najjar S, Pearlman D, Zagzag D, Devinsky O: resolução espontânea
encefalite limbica auto-imune seronegativa. Cogn Behav Neurol 2011,
24: 99 105.
114. Gabilondo I, Saiz A, Galan L, Gonzalez V, Jadraque R, Sabater L, Sans A,
Sempere A, Vela A, Villalobos F, Vi als M, Villoslada P, Graus F: Análise de
recidivas na encefalite anti-NMDAR. Neurology 2011, 77: 996 999.
115. Barry H, Hardiman O, Healy DG, Keogan M, Moroney J, Molnar PP, Cotter
DR, Murphy KC: encefalite do receptor anti-NMDA: um importante
diagnóstico diferencial em psicose. Br J Psychiatry 2011, 199: 508 509.
116. Dickerson F, Stallings C, Vaughan C, Origoni A, Khushalani S, Yolken R:
Anticorpos para o receptor de glutamato na mania. Bipolar Disord 2012,
14: 547 553.
117. O'Loughlin K, Ruge P, McCauley M: Encefalite e esquizofrenia: a
questão de palavras. Br J Psychiatry 2012, 201: 74.
118. Parratt KL, Allan M, Lewis SJ, Dalmau J, Halmagyi GM, Spies JM: aguda
Doença psiquiátrica em uma jovem mulher: uma forma incomum de encefalite.
Med J Aust 2009, 191: 284 286.
119. Suzuki Y, Kurita T, Sakurai K, Takeda Y, Koyama T: Relato de caso de anti-NMDA
encefalite receptora suspeita de esquizofrenia. Seishin Shinkeigaku
Zasshi 2009, 111: 1479 1484.
120. Tsutsui K, Kanbayashi T, Tanaka K, Boku S, Ito W, Tokunaga J, Mori A,
Hishikawa Y, Shimizu T, Nishino S: anticorpo anti-receptor de NMDA detectado
na encefalite, esquizofrenia e narcolepsia com características psicóticas.
BMC Psychiatry 2012, 12: 37.
121. Van Putten WK, Hachimi-Idrissi S, Jansen A, Van Gorp V, Huyghens L:
Causa incomum de comportamento psicótico em uma menina 9 de um ano: um caso
relatório. Relatório de caso Med 2012, 2012: 358520.
122. Masdeu JC, Gonzalez-Pinto A, Matute C, Ruiz De Azua S, Palomino A, De
Leon J, Berman KF, Dalmau J: anticorpos IgG de soro contra o NR1
subunidade do receptor NMDA não detectado na esquizofrenia. Am J
Psychiatry 2012, 169: 1120 1121.
123. Kirvan CA, Swedo SE, Kurahara D, Cunningham MW: mimetismo estreptocócico
e a sinalização celular mediada por anticorpos na patogênese de
Coréia de Sydenham. Autoimmunity 2006, 39: 21 29.
124. Swedo SE: infecção estreptocócica, síndrome de Tourette e TOC: existe
uma conexão? Pandas: Cavalo ou zebra? Neurology 2010, 74: 1397 1398.
125. Morer A, Lazaro L, Sabater L, Massana J, Castro J, Graus F: Antineuronal
anticorpos em um grupo de crianças com transtorno obsessivo-compulsivo
e síndrome de Tourette. J Psychiatr Res 2008, 42: 64 68.
126. Pavone P, Bianchini R, Parano E, Incorpora G, Rizzo R, Mazzone L, Trifiletti RR:
Anticorpos anti-cérebro em PANDAS versus estreptococo não complicado
infecção. Pediatr Neurol 2004, 30: 107 110.
127. Maina G, Albert U, Bogetto F, Borghese C, Berro AC, Mutani R, Rossi F,
Vigliani MC: anticorpos anti-cérebro em pacientes adultos com obsessivecompulsivo
transtorno. J Affect Disord 2009, 116: 192 200.
128. Brimberg L, Benhar I, Mascaro-Blanco A, Álvarez K, Lotan D, Inverno C, Klein J,
Moses AE, Somnier FE, Leckman JF, Swedo SE, Cunningham MW, Joel D:
Anormalidades comportamentais, farmacológicas e imunológicas após
exposição estreptocócica: um modelo de rato novo de Coreia de Sydenham e
distúrbios neuropsiquiátricos relacionados. Neuropsicofarmacologia 2012,
37: 2076 2087.
129. Dale RC, Candler PM, Church AJ, Wait R, Pocock JM, Giovannoni G:
As enzimas glicolíticas da superfície neuronal são alvos de autoantigénio em
doença pós-estreptocócica do SNC autoimune. J Neuroimmunol 2006,
172: 187 197.
130. Nicholson TR, Ferdinando S, Krishnaiah RB, Anhoury S, Lennox BR, MataixCols
D, Cleare A, Veale DM, Drummond LM, Fineberg NA, Igreja AJ,
Giovannoni G, Heyman I: prevalência de anticorpos anti gânglios anti-basais em
transtorno obsessivo-compulsivo do adulto: estudo transversal. Br J Psiquiatria
2012, 200: 381 386.
131. Wu K, Hanna GL, Rosenberg DR, Arnold PD: o papel do glutamato
sinalização na patogênese e tratamento de obsessivo-compulsivo
transtorno. Pharmacol Biochem Behav 2012, 100: 726 735.
132. Perlmutter SJ, Leitman SF, Garvey MA, Hamburger S, Feldman E, Leonard
HL, Swedo SE: troca de plasma terapêutico e intravenosa
imunoglobulina para transtorno obsessivo-compulsivo e distúrbios ticos em
infância. Lancet 1999, 354: 1153 1158.
133. Pereira A Jr, Furlan FA: Astrocytes e cognição humana: modelagem
integração de informações e modulação da atividade neuronal.
Prog Neurobiol 2010, 92: 405 420.
134. Barres BA: o mistério e a magia da glia: uma perspectiva sobre seus papéis em
saúde e doença. Neuron 2008, 60: 430 440.
135. Verkhratsky A, Parpura V, Rodriguez JJ: onde os pensamentos habitam: o
fisiologia da rede neural neuronal-glial "difusa". Brain Res Rev 2011,
66: 133 151.
136. Sofroniew MV: dissecção molecular de astrogliose reativa e cicatriz glial
formação. Trends Neurosci 2009, 32: 638 647.
137. Hamilton NB, Attwell D: Os astrocitos realmente exocitam neurotransmissores?
Nat Rev Neurosci 2010, 11: 227 238.
138. Rajkowska G: estudos pós-morte em distúrbios de humor indicam alteração
número de neurônios e células gliais. Biol Psychiatry 2000, 48: 766 777.
139. Coupland NJ, Ogilvie CJ, Hegadoren KM, Seres P, Hanstock CC, Allen PS:
Diminuição do Myo-inositol pré-frontal em transtorno depressivo maior.
Biol Psychiatry 2005, 57: 1526 1534.
140. Miguel-Hidalgo JJ, Overholser JC, Jurjus GJ, Meltzer HY, Dieter L, Konick L,
Stockmeier CA, Rajkowska G: imunorreatividade vascular e extravascular
para a molécula de adesão intercelular 1 no córtex orbitofrontal de
indivíduos com depressão maior: mudanças dependentes da idade. J Affect Disord
2011, 132: 422 431.
141. Miguel-Hidalgo JJ, Wei JR, Andrew M, Overholser JC, Jurjus G, Stockmeier
CA, Rajkowska G: patologia da glia no córtex pré-frontal em álcool
dependência com e sem sintomas depressivos. Biol Psiquiatria 2002,
52: 1121 1133.
142. Stockmeier CA, Mahajan GJ, Konick LC, Overholser JC, Jurjus GJ, Meltzer HY,
Uylings HB, Friedman L, Rajkowska G: Alterações celulares na pós-morte
hipocampo na depressão maior. Biol Psychiatry 2004, 56: 640 650.
143. Ong r D, Drevets WC, Price JL: Redução glial no subgenual pré-frontal
córtex nos transtornos de humor. Proc Natl Acad Sci USA 1998, 95: 13290 13295.
144. Gittins RA, Harrison PJ: estudo morfométrico de glia e neurônios na
córtex cingulado anterior no transtorno do humor. J Affect Disord 2011,
133: 328 332.
145. Cotter D, Mackay D, Beasley C, Kerwin R, Everall I: Densidade glial reduzida
e volume neuronal em transtorno depressivo maior e esquizofrenia em
córtex cingulado anterior [resumo]. Esquizofrenia Res 2000, 41: 106.
146. Si X, Miguel-Hidalgo JJ, Rajkowska G: a expressão de GFAP é reduzida no
córtex pré-frontal dorsolateral na depressão. Na Sociedade de Neurociências; 2003.
Reunião de Neurociências Planne: Nova Orleans; 2003.
147. Legutko B, Mahajan G, Stockmeier CA, Rajkowska G: Astrócitos de matéria branca
são reduzidos na depressão. Na Society for Neuroscience. Reunião de Neurociências
Planejador: Washington, DC; 2011.148. Edgar N, Sibille E: um papel funcional putativo para oligodendrócitos em
regulação do humor. Transl Psychiatry 2012, 2: e109.
149. Rajkowska G, Halaris A, Selemon LD: Reduções em neurônios e gliais
densidade caracteriza o córtex pré-frontal dorsolateral em bipolar
transtorno. Biol Psychiatry 2001, 49: 741 752.
150. Cotter D, Mackay D, Chana G, Beasley C, Landau S, Everall IP: Reduzida
tamanho neuronal e densidade celular glial na área 9 do dorsolateral
córtex pré-frontal em indivíduos com transtorno depressivo maior. Cereb Cortex
2002, 12: 386 394.
151. Stark AK, Uylings HB, Sanz-Arigita E, Pakkenberg B: perda de células gliales no
córtex cingulado anterior, uma sub-região do córtex pré-frontal, em
sujeitos com esquizofrenia. Am J Psychiatry 2004, 161: 882 888.
152. Konopaske GT, Dorph-Petersen KA, Sweet RA, Pierri JN, Zhang W, Sampson
AR, Lewis DA: efeito da exposição antipsicótica crônica em astrocitos e
números de oligodendrócitos em macacos macacos. Biol Psiquiatria 2008,
63: 759 765.
153. Selemon LD, Lidow MS, Goldman-Rakic PS: Aumento de volume e glial
densidade no córtex pré-frontal de primatas associada a crônica
exposição a medicamentos antipsicóticos. Biol Psychiatry 1999, 46: 161 172.
154. Steiner J, Bernstein HG, Bielau H, Farkas N, Winter J, Dobrowolny H, Brisch R,
Gos T, Mawrin C, Myint AM, Bogerts B: S100B-glia imunopositiva é
elevado em paranóico em comparação com a esquizofrenia residual: a
estudo morfométrico. J Psychiatr Res 2008, 42: 868 876.
155. Carter CJ: eIF2B e sobrevivência de oligodendrócitos: onde a natureza e nutrição
Encontra-se no transtorno bipolar e na esquizofrenia? Schizophr Bull 2007,
33: 1343 1353.
156. Hayashi Y, Nihonmatsu-Kikuchi N, Hisanaga S, Yu XJ, Tatebayashi Y:
Neuropatologia semelhanças e diferenças entre esquizofrenia
e transtorno bipolar: um estudo de córtex pós-morte morto de fluxo.
PLoS One 2012, 7: e33019.
157. Uranova NA, Vikhreva OV, Rachmanova VI, Orlovskaya DD: Ultrastructural
alterações de fibras mielinizadas e oligodendrócitos no pré-frontal
Córtex na esquizofrenia: um estudo morfométrico pós-mortem.
Schizophr Res Tratamento 2011, 2011: 325789.
158. Torres-Platas SG, Hercher C, Davoli MA, Maussion G, Labonte B, Turecki
G, Mechawar N: hipertrofia astrocítica no branco cingulado anterior
assunto de suicídios deprimidos. Neuropsicofarmacologia 2011,
36: 2650 2658.
159. Pereira A Jr, Furlan FA: Sobre o papel da sincronia para os neurônios-astrócitos
interações e processamento perceptual consciente. J Biol Phys 2009,
35: 465 480.
160. Kettenmann H, Hanisch Reino Unido, Noda M, Verkhratsky A: Fisiologia de
microglia. Physiol Rev 2011, 91: 461 553.
161. Tremblay ME, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A: o papel
de microglia no cérebro saudável. J Neurosci 2011, 31: 16064 16069.
162. Kaindl AM, Degos V, Peineau S, Gouadon E, Chhor V, Loron G, Le
Charpentier T, Josserand J, Ali C, Vivien D, Collingridge GL, Lombet A, Issa L,
Rene F, Loeffler JP, Kavelaars A, Verney C, Mantz J, Gressens P: Ativação de
Os receptores microgliais de N-metil-D-aspartato desencadeiam inflamação e
morte celular neuronal no cérebro em desenvolvimento e maduro. Ann Neurol
2012, 72: 536 549.
163. Schwartz M, Shaked I, Fisher J, Mizrahi T, Schori H: Protetor
Auto-imunidade contra o inimigo dentro: combate à toxicidade do glutamato.
Trends Neurosci 2003, 26: 297 302.
164. Pacheco R, Gallart T, Lluis C, Franco R: Papel do glutamato na célula T
imunidade mediada. J Neuroimmunol 2007, 185: 9 19.
165. Najjar S, Pearlman D, Miller DC, Devinsky O: epilepsia refratária associada
com ativação microglial. Neurologist 2011, 17: 249 254.
166. Schwartz M, Butovsky O, Bruck W, Hanisch Reino Unido: fenótipo microglial: é o
compromisso reversível? Trends Neurosci 2006, 29: 68 74.
167. Wang F, Wu H, Xu S, Guo X, Yang J, Shen X: migração de macrófagos
o fator inibidor activa a ciclooxigenase 2-prostaglandina E2 em cultura
microglia espinhal. Neurosci Res 2011, 71: 210 218.
168. Zhang XY, Xiu MH, Song C, Chenda C, Wu GY, Haile CN, Kosten TA, Kosten
TR: Aumento do S100B no soro nunca medicado e medicado
pacientes esquizofrênicos. J Psychiatr Res 2010, 44: 1236 1240.
169. Kawasaki Y, Zhang L, Cheng JK, Ji RR: mecanismos de citoquinas da central
sensibilização: papel distinto e sobreposto da interleucina-1beta,
interleucina-6 e factor de necrose tumoral alfa na regulação sináptica e
atividade neuronal na medula espinhal superficial. J Neurosci 2008,
28: 5189 5194.
170. M ller N, Schwarz MJ: The immunological basis of glutamatérgic
perturbação na esquizofrenia: para uma visão integrada. J Neural
Transm Suppl 2007, 72: 269 280.
171. Hestad KA, Tonseth S, Stoen CD, Ueland T, Aukrust P: níveis plasmáticos aumentados
do fator de necrose tumoral alfa em pacientes com depressão: normalização
durante a terapia eletroconvulsiva. J ECT 2003, 19: 183 188.
172. Kubera M, Kenis G, Bosmans E, Zieba A, Dudek D, Nowak G, Maes M:
Níveis plasmáticos de interleucina-6, interleucina-10 e receptor interleucina-1
antagonista na depressão: comparação entre o estado agudo e após
remissão. Pol J Pharmacol 2000, 52: 237 241.
173. Miller BJ, Buckley P, Seabolt W, Mellor A, Kirkpatrick B: Meta-análise de
Alterações de citoquinas na esquizofrenia: estado clínico e antipsicótico
efeitos. Biol Psychiatry 2011, 70: 663 671.
174. Potvin S, Stip E, Sepehry AA, Gendron A, Bah R, Kouassi E: Inflamatórios
Alterações citocinas na esquizofrenia: uma revisão quantitativa sistemática.
Biol Psychiatry 2008, 63: 801 808.
175. Reale M, Patruno A, De Lutiis MA, Pesce M, Felaco M, Di Giannantonio M, Di
Nicola M, Grilli A: Disregulação da produção de quimiocitocina em
pacientes esquizofrênicos versus controles saudáveis. BMC Neurosci 2011, 12: 13.
176. Fluitman SB, Denys DA, Heijnen CJ, Westenberg HG: O desgosto afeta TNFalfa,
Níveis de IL-6 e noradrenalina em pacientes com obsessivo-compulsivo
transtorno. Psychoneuroendocrinology 2010, 35: 906 911.
177. Konuk N, Tekin IO, Ozturk U, Atik L, Atasoy N, Bektas S, Erdogan A: Plasma
níveis de factor de necrose tumoral alfa e interleucina-6 em obsessivo
desordem compulsiva. Mediadores Inflamm 2007, 2007: 65704.
178. Monteleone P, Catapano F, Fabrazzo M, Tortorella A, Maj M: Diminuição
níveis sanguíneos de factor de necrose tumoral alfa em pacientes com obsessivecompulsivo
transtorno. Neuropsychobiology 1998, 37: 182 185.
179. Marazziti D, Presta S, Pfanner C, Gemignani A, Rossi A, Sbrana S, Rocchi V,
Ambrogi F, Cassano GB: alterações imunológicas em adultos obsessivecompulsivas
transtorno. Biol Psychiatry 1999, 46: 810 814.
180. Zai G, Arnold PD, Burroughs E, Richter MA, Kennedy JL: necrose tumoral
O gene factor-alfa não está associado a transtorno obsessivo-compulsivo.
Psychiatr Genet 2006, 16: 43.
181. Rodr guez AD, Gonz lez PA, Garc a MJ, de la Rosa A., Vargas M, Marrero F:
Variações circadianas nas concentrações de citocinas pró-inflamatórias em aguda
infarto do miocárdio. Rev Esp Cardiol 2003, 56: 555 ± 560.
182. Oliver JC, Bland LA, Oettinger CW, Arduino MJ, McAllister SK, Aguero SM,
Favero MS: Cicloquina cinética em um modelo in vitro de sangue total seguindo
um desafio de endotoxina. Lymphokine Cytokine Res 1993, 12: 115 ± 120.
183. Le T, Leung L, Carroll WL, Schibler KR: Regulação do gene interleucina-10
expressão: mecanismos possíveis que contabilizam sua upregulation e para
diferenças maturacionais na sua expressão por células mononucleares no sangue.
Blood 1997, 89: 4112 4119.
184. Lee MC, Ting KK, Adams S, Brew BJ, Chung R, Guillemin GJ:
Caracterização da expressão de receptores NMDA em humanos
astrocitos. PLoS One 2010, 5: e14123.
185. Myint AM, Kim YK, Verkerk R, Scharpe S, Steinbusch H, Leonard B:
Via kynurenina na depressão maior: evidência de deficiência
neuroproteção. J Affect Disord 2007, 98: 143 151.
186. Sanacora G, Treccani G, Popoli M: Rumo a uma hipótese de glutamato de
depressão: uma fronteira emergente da neuropsicofarmacologia para
Transtornos de Humor. Neuropharmacology 2012, 62: 63 77.
187. Saleh A, Schroeter M, Jonkmanns C, Hartung HP, Modder U, Jander S: Em
MRI viral de inflamação cerebral no acidente vascular cerebral isquêmico humano. Brain 2004,
127: 1670 1677.
188. Tilleux S, Hermans E: Neuroinflamação e regulação do glutamato glial
captação em distúrbios neurológicos. J Neurosci Res 2007, 85: 2059 ± 2070.
189. Helms HC, Madelung R, Waagepetersen HS, Nielsen CU, Brodin B: In vitro
evidência para a hipótese do refluxo do glutamato cerebral: endotélio cerebral
As células coctivadas com astrocitos apresentam um cérebro polarizado
transporte de glutamato. 2012, 60: 882 893.
190. Leonard BE: o conceito de depressão como uma disfunção do sistema imunológico
sistema. Curr Immunol Rev 2010, 6: 205 212.
191. Labrie V, Wong AH, Roder JC: Contribuições da via D-serina para
esquizofrenia. Neuropharmacology 2012, 62: 1484 1503.
192. Gras G, Samah B, Hubert A, Leone C, Porcheray F, Rimaniol AC: EAAT
expressão por macrófagos e microglia: ainda mais perguntas do que
respostas. Amino Acids 2012, 42: 221 229.
193. Livingstone PD, Dickinson JA, Srinivasan J, Kew JN, Wonnacott S:
O crosstalk de glutamato-dopamina no córtex pré-frontal de ratos é modulado pelos receptores nicotínicos Alpha7 e potencializado por PNU-120596. J Mol
Neurosci 2010, 40: 172 176.194. Kondziella D, Brenner E, Eyjolfsson EM, Sonnewald U: How do glialneuronal
as interações se encaixam nas atuais hipóteses de neurotransmissores de
esquizofrenia? Neurochem Int 2007, 50: 291 301.
195. Wu HQ, Pereira EF, Bruno JP, Pellicciari R, Albuquerque EX, Schwarcz R: O
antagonista do receptor nicotínico alfa7 derivado de astrocitos ácido kynurenic
controla os níveis extracelulares de glutamato no córtex pré-frontal. J Mol
Neurosci 2010, 40: 204 210.
196. Steiner J, Bogerts B, Schroeter ML, Bernstein HG: proteína S100B em
doenças neurodegenerativas. Clin Chem Lab Med 2011, 49: 409 424.
197. Steiner J, Marquardt N, Pauls I, Schiltz K, Rahmoune H, Bahn S, Bogerts B,
Schmidt RE, Jacobs R: células CD8 humanas (+) e células NK expressam e
secretar S100B após estimulação. Brain Behav Immun 2011, 25: 1233 1241.
198. Shanmugam N, Kim YS, Lanting L, Natarajan R: Regulamento de
expressão de ciclooxigenase-2 em monócitos por ligação do receptor
para produtos finais de glicação avançada. J Biol Chem 2003, 278: 34834 34844.
199. Rothermundt M, Ohrmann P, Abel S, Siegmund A, Pedersen A, Ponath G,
Suslow T, Peters M, Kaestner F, Heindel W, Arolt V, Pfleiderer B: célula glial
ativação em um subgrupo de pacientes com esquizofrenia indicada por
aumento das concentrações séricas de S100B e mio-inositol elevado.
Prog Neuropsychopharmacol Biol Psychiatry 2007, 31: 361 364.
200. Falcone T, Fazio V, Lee C, Simon B, Franco K, Marchi N, Janigro D: Soro
S100B: um potencial biomarcador para suicídio em adolescentes? PLoS One
2010, 5: e11089.
201. Schroeter ML, Abdul-Khaliq H, Krebs M, Diefenbacher A, Blasig IE: soro
Os marcadores suportam a patologia glial específica da doença na depressão maior.
J Affect Disord 2008, 111: 271 280.
202. Rothermundt M, Ahn JN, Jorgens S: S100B em esquizofrenia: uma atualização.
Gen Physiol Biophys 2009, 28 Spec No Focus: F76 F81.
203. Schroeter ML, Abdul-Khaliq H, Krebs M, Diefenbacher A, Blasig IE: Neurônico
a enolase é inalterada enquanto a S100B está elevada no soro de
pacientes com esquizofrenia - pesquisa original e meta-análise.
Psychiatry Res 2009, 167: 66 72.
204. Rothermundt M, Missler U, Arolt V, Peters M, Leadbeater J, Wiesmann M,
Rudolf S, Wandinger KP, Kirchner H: Aumento dos níveis sanguíneos de S100B em
pacientes esquizofrênicos não medicados e tratados estão correlacionados com
sintomatologia negativa. Mol Psychiatry 2001, 6: 445 449.
205. Suchankova P, Klang J, Cavanna C, Holm G, Nilsson S, Jonsson EG, Ekman A:
O polimorfismo Gly82Ser no gene RAGE é relevante para
esquizofrenia e psicopatia do traço de personalidade? J Psychiatry Neurosci
2012, 37: 122 128.
206. Scapagnini G, Davinelli S, Drago F, De Lorenzo A, Oriani G: Antioxidantes como
antidepressivos: fato ou ficção? CNS Drugs 2012, 26: 477 490.
207. Ng F, Berk M, Dean O, Bush AI: estresse oxidativo em transtornos psiquiátricos:
base de evidências e implicações terapêuticas. Int J Neuropsychopharmacol
2008, 11: 851 876.
208. Salim S, Chugh G, Asghar M: inflamação na ansiedade. Adv Protein Chem
Struct Biol 2012, 88: 1 25.
209. Anderson G, Berk M, Dodd S, Bechter K, Altamura AC, Dell'osso B, Kanba S,
Monji A, Fatemi SH, Buckley P, Debnath M, Das UN, Meyer U, M ller N,
Kanchanatawan B, Maes M: imuno-inflamatório, oxidativo e nitrosativo
estresse e caminhos neuroprogressivos na etiologia, curso e tratamento
da esquizofrenia. Prog Neuropsychopharmacol Biol Psychiatry 2013, 42: 1 42.
210. Coughlin JM, Ishizuka K, Kano SI, Edwards JA, Seifuddin FT, Shimano MA,
Daley EL, et al: redução marcada de superóxido dismutase solúvel-1
(SOD1) no líquido cefalorraquidiano de pacientes com início recente
esquizofrenia. Mol Psychiatry 2012, 18: 10 11.
211. Bombaci M, Grifantini R, Mora M, Reguzzi V, Petracca R, Meoni E, Balloni S,
Zingaretti C, Falugi F, Manetti AG, Margarit I, Musser JM, Cardona F, Orefici
G, Grandi G, Bensi G: O perfil da matriz de proteínas dos soros do paciente tic revela um
ampla gama e resposta imune melhorada contra o Grupo A
Antígenos de Streptococcus. PLoS One 2009, 4: e6332.
212. Valerio A, Cardile A, Cozzi V, Bracale R, Tedesco L, Pisconti A, Palomba L,
Cantoni O, Clementi E, Moncada S, Carruba MO, Nisoli E: TNF-alfa
downregulates eNOS expressão e biogênese mitocondrial em gordura
e músculos de roedores obesos. J Clin Invest 2006, 116: 2791 2798.
213. Ott M, Gogvadze V, Orrenius S, Zhivotovsky B: mitocôndrias, oxidativas
estresse e morte celular. Apoptosis 2007, 12: 913 922.
214. Shalev H, Serlin Y, Friedman A: Romper a barreira hematoencefálica como um portão
para transtorno psiquiátrico. Cardiovasc Psiquiatria Neurol 2009, 2009: 278531.
215. Abbott NJ, Ronnback L, Hansson E: interações endotélias astrocitas na
barreira hematoencefálica. Nat Rev Neurosci 2006, 7: 41 53.
216. Bechter K, Reiber H, Herzog S, Fuchs D, Tumani H, Maxeiner HG:
Análise do líquido cefalorraquidiano no espectro afetivo e esquizofrênico
Distúrbios: identificação de subgrupos com respostas imunes e
disfunção da barreira hematoencefálica. J Psychiatr Res 2010, 44: 321 330.
217. Harris LW, Wayland M, Lan M, Ryan M, Giger T, Lockstone H, Wuethrich I,
Mimmack M, Wang L, Kotter M, Craddock R, Bahn S: O cerebral
microvasculatura em esquizofrenia: um estudo de microdissecão de captura a laser.
PLoS One 2008, 3: e3964.
218. Lin JJ, Mula M, Hermann BP: Descobrindo o neurobehavioural
comorbidades da epilepsia ao longo da vida. Lancet 2012, 380: 1180 1192.
219. Isingrini E, Belzung C, Freslon JL, Machet MC, Camus V: efeito de fluoxetina sobre
Vasorelaxação dependente de óxido nítrico aórtico no imprevisível
Modelo de estresse leve crônico de depressão em camundongos. Psychosom Med 2012,
74: 63 72.
220. Zhang XY, Zhou DF, Cao LY, Zhang PY, Wu GY, Shen YC: O efeito de
tratamento com risperidona em superóxido dismutase na esquizofrenia. J Clin
Psychopharmacol 2003, 23: 128 131.
221. Lavoie KL, Pelletier R, Arsenault A, Dupuis J, Bacon SL: associação entre
depressão clínica e função endotelial medida pelo antebraço
reatividade hiperêmica. Psychosom Med 2010, 72: 20 26.
222. Chrapko W, Jurasz P, Radomski MW, Archer SL, Newman SC, Baker G, Lara N,
Le Melledo JM: alteração da diminuição dos metabolitos de plasma NO e
atividade plaquetária de NO sintase por paroxetina em pacientes deprimidos.
Neuropsychopharmacology 2006, 31: 1286 1293.
223. Chrapko WE, Jurasz P, Radomski MW, Lara N, Archer SL, Le Melledo JM:
Diminuição da atividade das plaquetas de óxido nítrico sintase e do óxido nítrico plasmático
metabólitos no transtorno depressivo maior. Biol Psychiatry 2004, 56: 129 134.
224. Stuehr DJ, Santolini J, Wang ZQ, Wei CC, Adak S: Atualização sobre o mecanismo
e regulação catalítica nas NO sintases. J Biol Chem 2004,
279: 36167 36170.
225. Chen W, Druhan LJ, Chen CA, Hemann C, Chen YR, Berka V, Tsai AL, Zweier
JL: o peroxinitrito induz a destruição da tetrahidrobiopterina e
Heme em óxido nítrico sintase endotelial: transição de reversível para
inibição enzimática irreversível. Biochemistry 2010, 49: 3129 3137.
226. Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA, Chen
YR, Druhan LJ, Zweier JL: S-glutationilação desacopla eNOS e
regula sua função celular e vascular. Nature 2010, 468: 1115 1118.
227. Szabo C, Ischiropoulos H, Radi R: Peroxinitrito: bioquímica,
fisiopatologia e desenvolvimento de terapêutica. Nat Rev Drug Discov
2007, 6: 662 680.
228. Papakostas GI, Shelton RC, Zajecka JM, Etemad B, Rickels K, Clain A, Baer L,
Dalton ED, Sacco GR, Schoenfeld D, Pencina M, Meisner A, Bottiglieri T,
Nelson E, Mischoulon D, Alpert JE, Barbee JG, Zisook S, Fava M: Lmetilfolato
como terapia adjuvante para depressão maior resistente a SSRI:
resultados de dois ensaios randomizados, em dupla ocultação, em paralelo sequencial. Am J
Psychiatry 2012, 169: 1267 1274.
229. Antoniades C, Shirodaria C, Warrick N, Cai S, de Bono J, Lee J, Leeson P,
Neubauer S, Ratnatunga C, Pillai R, Refsum H, Channon KM: 5-
metiltetrahidrofolato rapidamente melhora a função endotelial e
diminui a produção de superóxido em vasos humanos: efeitos sobre vasos
disponibilidade de tetrahidrobiopterina e sintetase de óxido nítrico endotelial
acoplamento. Circulation 2006, 114: 1193 1201.
230. Masano T, Kawashima S, Toh R, Satomi-Kobayashi S, Shinohara M, Takaya T,
Sasaki N, Takeda M, Tawa H, Yamashita T, Yokoyama M, Hirata K: Beneficiário
Efeitos da tetrahidrobiopterina exógena na remodelação do ventrículo esquerdo
após infarto do miocárdio em ratos: o possível papel do estresse oxidativo
causada pela sintase de óxido nítrico endotelial desacoplada. Circ J 2008,
72: 1512 1519.
231. Alp NJ, Channon KM: regulação da óxido nítrico sintase endotelial por
tetrahidrobiopterina em doença vascular. Arterioscler Thromb Vasc Biol 2004,
24: 413 420.
232. Szymanski S, Ashtari M, Zito J, Degreef G, Bogerts B, Lieberman J:
Escalas de ressonância magnética de eco de gradiente aumentado de Gadolinium-DTPA em
primeiro episódio de psicose e pacientes esquizofrênicos crônicos.
Psychiatry Res 1991, 40: 203 207.
233. Butler T, Weisholtz D, Isenberg N, Harding E, Epstein J, Stern E, Silbersweig
D: Neuroimagem da disfunção frontal-limbica na esquizofrenia e
psicose relacionada à epilepsia: para uma neurobiologia convergente.
Epilepsy Behav 2012, 23: 113 122.234. Butler T, Maoz A, Vallabhajosula S, Moeller J, Ichise M, Paresh K, Pervez F,
Friedman D, Goldsmith S, Najjar S, Osborne J, Solnes L, Wang X, francês J,
Thesen T, Devinsky O, Kuzniecky R, Stern E, Silbersweig D: Imaging
inflamação em um paciente com epilepsia associada a anticorpos para
ácido glutamico descarboxilase [resumo]. Em Am Epilepsy Society Abstracts,
Volume 2. Baltimore: American Epilepsy Society; 2011: 191.
235. van Berckel BN, Bossong MG, Boellaard R, Kloet R, Schuitemaker A, Caspers
E, Luurtsema G, Windhorst AD, Cahn W, Lammertsma AA, Kahn RS:
Ativação de microglia em esquizofrenia de início recente: um (R) -
[11C] estudo de tomografia por emissão de pósitrons PK11195. Biol Psiquiatria 2008,
64: 820 822.
236. Doorduin J, de Vries EF, Willemsen AT, de Groot JC, Dierckx RA, Klein HC:
Neuroinflamação na psicose relacionada à esquizofrenia: um estudo de PET.
J Nucl Med 2009, 50: 1801 1807.
237. Takano A, Arakawa R, Ito H, Tateno A, Takahashi H, Matsumoto R, Okubo Y,
Suhara T: receptores periféricos de benzodiazepina em pacientes com
esquizofrenia: um estudo de PET com [11C] DAA1106. Int J
Neuropsychopharmacol 2010, 13: 943 ± 950.
238. M ller N, Schwarz MJ, Dehning S, Douhe A, Cerovecki A, Goldstein-Muller B,
Spellmann I, Hetzel G, Maino K, Kleindienst N, M ller HJ, Arolt V, Riedel M:
O inibidor de ciclooxigenase-2 celecoxib tem efeitos terapêuticos em
depressão maior: resultados de um placebo duplo-cego, aleatorizado
controlado, estudo piloto adicional para reboxetina. Mol Psychiatry 2006,
11: 680 684.
239. Akhondzadeh S, Jafari S, Raisi F, Nasehi AA, Ghoreishi A, Salehi B, MohebbiRasa
S, Raznahan M, Kamalipour A: Ensaio clínico de celecoxib adjuvante
tratamento em pacientes com depressão maior: um duplo cego e
ensaio controlado com placebo. Depress Anxiety 2009, 26: 607 611.
240. Mendlewicz J, Kriwin P, Oswald P, Souery D, Alboni S, Brunello N:
Acúmulo acentuado de ação de antidepressivos na depressão maior usando
Aumento de ácido acetilsalicílico: um estudo piloto aberto. Int Clin
Psychopharmacol 2006, 21: 227 231.
241. Uher R, Carver S, Power RA, Mors O, Maier W, Rietschel M, Hauser J,
Dernovsek MZ, Henigsberg N, Souery D, Placentino A, Agricultor A, McGuffin P:
Medicamentos anti-inflamatórios não esteróides e eficácia de antidepressivos em
transtorno depressivo maior. Psychol Med 2012, 42: 2027 2035.
242. M ller N, Riedel M, Scheppach C, Brandstatter B, Sokullu S, Krampe K,
Ulmschneider M, Engel RR, Moller HJ, Schwarz MJ: antipsicótico benéfico
efeitos da terapia complementar de celecoxib em comparação com a risperidona isolada em
esquizofrenia. Am J Psychiatry 2002, 159: 1029 1034.
243. M ller N, Riedel M, Schwarz MJ, Engel RR: Clinical effects of COX-2
Inibidores da cognição na esquizofrenia. Eur Arch Psychiatry Clin Neurosci
2005, 255: 149 151.
244. M ller N, Krause D, Dehning S, Musil R, Schennach-Wolff R, Obermeier M,
Moller HJ, Klauss V, Schwarz MJ, Riedel M: tratamento com Celecoxib no início
estágio de esquizofrenia: resultados de um estudo randomizado, duplo-cego, placebocontrolled
Ensaio do aumento de celecoxib do tratamento com amisulprida.
Schizophr Res 2010, 121: 118 124.
245. Sayyah M, Boostani H, Pakseresht S, Malayeri A: um randomizado preliminar
ensaio clínico duplo-cego sobre a eficácia do celecoxib como adjuvante na
o tratamento do transtorno obsessivo-compulsivo. Psiquiatria Res 2011,
189: 403 406.
246. Sublette ME, Ellis SP, Geant AL, Mann JJ: Meta-análise dos efeitos de
Ácido eicosapentaenóico (EPA) em ensaios clínicos em depressão. J Clin
Psychiatry 2011, 72: 1577 1584.
247. Bloch MH, Hannestad J: Omega-3 ácidos graxos para o tratamento de
depressão: revisão sistemática e meta-análise. Mol Psychiatry 2012,
17: 1272 1282.
248. Keller WR, Kum LM, Wehring HJ, Koola MM, Buchanan RW, Kelly DL: A
revisão de agentes anti-inflamatórios para sintomas de esquizofrenia.
J Psychopharmacol.
249. Warner-Schmidt JL, Vanover KE, Chen EY, Marshall JJ, Greengard P:
Efeitos antidepressivos dos inibidores seletivos da recaptação da serotonina (SSRIs)
são atenuados por drogas anti-inflamatórias em camundongos e humanos. Proc Natl
Acad Sci USA 2011, 108: 9262 9267.
250. Gallagher PJ, Castro V, Fava M, Weilburg JB, Murphy SN, Gainer VS, Churchill
SE, Kohane IS, Iosifescu DV, Smoller JW, Perlis RH: resposta antidepressiva
em pacientes com depressão maior expostos a AINEs: um
estudo de farmacovigilância. Am J Psychiatry 2012, 169: 1065 1072.
251. Shelton RC: O uso concomitante de AINEs reduz a eficácia de
antidepressivos? Am J Psychiatry 2012, 169: 1012 1015.
252. Martinez-Gras I, Perez-Nievas BG, Garcia-Bueno B, Madrigal JL, AndresEsteban
E, Rodriguez-Jimenez R, Hoenicka J, Palomo T, Rubio G, Leza JC:
A prostaglandina anti-inflamatória 15d-PGJ2 e seu receptor nuclear
PPARgamma são diminuídos na esquizofrenia. Schizophr Res 2011,
128: 15 22.
253. Garcia-Bueno B, Perez-Nievas BG, Leza JC: Existe um papel para o nuclear
receptor PPARgamma em doenças neuropsiquiátricas? Int J
Neuropsychopharmacol 2010, 13: 1411 ± 1429.
254. Meyer U: Sinalização antiinflamatória na esquizofrenia. Brain Behav
Immun 2011, 25: 1507 1518.
255. Ramer R, Heinemann K, Merkord J, Rohde H, Salamon A, Linnebacher M,
Hinz B: COX-2 e PPAR-gama conferem apoptose induzida por cannabidiol
de células de câncer de pulmão humano. Mol Cancer Ther 2013, 12: 69 82.
256. Henry CJ, Huang Y, Wynne A, Hanke M, Himler J, Bailey MT, Sheridan JF,
Godbout JP: Minociclina atenua o lipopolisacarídeo (LPS) induzido
neuroinflamação, comportamento da doença e anedonia.
J Neuroinflamação 2008, 5: 15.
257. Sarris J, Mischoulon D, Schweitzer I: Omega-3 para transtorno bipolar: metaanalyses
de uso em mania e depressão bipolar. J Clin Psychiatry 2012,
73: 81 86.
258. Amminger GP, Schafer MR, Papageorgiou K, Klier CM, Cotton SM, Harrigan
SM, Mackinnon A, McGorry PD, Berger GE: ácidos graxos de cadeia longa omega-3
para prevenção recomendada de distúrbios psicóticos: um estudo randomizado, placebocontrolled
tentativas. Arch Gen Psychiatry 2010, 67: 146 154.
259. Fusar-Poli P, Berger G: intervenções de ácido eicosapentaenóico em
esquizofrenia: meta-análise de estudos randomizados e controlados por placebo.
J Clin Psychopharmacol 2012, 32: 179 ± 185.
260. Zorumski CF, Paul SM, Izumi Y, Covey DF, Mennerick S: Neurosteróides,
Estresse e depressão: Possíveis oportunidades terapêuticas.
Neurosci Biobehav Rev 2013, 37: 109 122.
261. Uhde TW, Singareddy R: Pesquisa Biológica em Distúrbios de Ansiedade. Dentro
Psiquiatria como uma Neurociência. Editado por Juan Jose LI, Wolfgang G, Mario M,
Norman S. Chichester: John Wiley & Sons, Ltd; 2002: 237 286.
262. Gibson SA, Korado Z, Shelton RC: estresse oxidativo e glutationa
resposta em culturas de tecidos de pessoas com depressão maior.
J Psychiatr Res 2012, 46: 1326 1332.
263. Nery FG, Monkul ES, Hatch JP, Fonseca M, Zunta-Soares GB, Frey BN,
Bowden CL, Soares JC: Celecoxib como adjuvante no tratamento de
episódios depressivos ou mistos de transtorno bipolar: um duplo cego,
estudo randomizado controlado com placebo. 2008, 23: 87 94.
264. Levine J, Cholestoy A, Zimmerman J: Possível efeito antidepressivo de
minociclina. 1996, 153: 582.
265. Levkovitz Y, Mendlovich S, Riwkes S, Braw Y, Levkovitch-Verbin H, Gal G,
Fennig S, Treves I, Kron S: estudo duplo-cego e aleatorizado de
minociclina para o tratamento de sintomas negativos e cognitivos em
esquizoprenia de fase inicial. J Clin Psychiatry 2010, 71: 138 149.
266. Miyaoka T, Yasukawa R, Yasuda H, Hayashida M, Inagaki T, Horiguchi J:
Possível efeito antipsicótico da minociclina em pacientes com
esquizofrenia. Prog Neuropsychopharmacol Biol Psychiatry 2007, 31: 304 307.
267. Miyaoka J, Yasukawa R, Yasuda H, Hayashida M, Inagaki T, Horiguchi J:
Minociclina como terapia adjuvante para esquizofrenia: uma etiqueta aberta
estudar. 2008, 31: 287 292.
268. Rodriguez CI, Bender J Jr, Marcus SM, Snape M, Rynn M, Simpson HB:
Aumento da farmacoterapia em Minociclina em obsessivo-compulsivo
desordem: um ensaio clínico aberto. 2010, 71: 1247 1249.
doi:10.1186/1742-2094-10-43
Cite este artigo como: Najjar et al .: Neuroinflamatório e psiquiátrico
doença. Jornal de Neuroinflamação 2013 10: 43.
Acordeão próximo
Escopo de prática profissional *
As informações aqui contidas em "Neuroinflamação e doença psiquiátrica" não se destina a substituir um relacionamento individual com um profissional de saúde qualificado ou médico licenciado e não é um conselho médico. Incentivamos você a tomar decisões de saúde com base em sua pesquisa e parceria com um profissional de saúde qualificado.
Informações do blog e discussões de escopo
Nosso escopo de informações limita-se à Quiropraxia, musculoesquelética, medicamentos físicos, bem-estar, contribuindo distúrbios viscerossomáticos dentro de apresentações clínicas, dinâmica clínica de reflexo somatovisceral associada, complexos de subluxação, questões de saúde sensíveis e/ou artigos, tópicos e discussões de medicina funcional.
Nós fornecemos e apresentamos colaboração clínica com especialistas de várias disciplinas. Cada especialista é regido por seu escopo profissional de prática e sua jurisdição de licenciamento. Usamos protocolos funcionais de saúde e bem-estar para tratar e apoiar o cuidado de lesões ou distúrbios do sistema músculo-esquelético.
Nossos vídeos, postagens, tópicos, assuntos e insights abrangem assuntos clínicos, problemas e tópicos relacionados e apoiam direta ou indiretamente nosso escopo de prática clínica.*
Nosso escritório tentou razoavelmente fornecer citações de apoio e identificou o estudo de pesquisa relevante ou estudos que apóiam nossas postagens. Fornecemos cópias dos estudos de pesquisa de apoio à disposição dos conselhos regulatórios e do público mediante solicitação.
Entendemos que cobrimos questões que requerem uma explicação adicional de como isso pode ajudar em um plano de cuidados ou protocolo de tratamento específico; portanto, para discutir melhor o assunto acima, sinta-se à vontade para perguntar Dr. Alex Jiménez, DC, ou contacte-nos 915-850-0900.
Estamos aqui para ajudar você e sua família.
Bênçãos
Dr. Alex Jimenez DC MSACP, RN*, CCST, IFMCP*, CIFM*, ATN*
o email: coach@elpasofunctionalmedicine. com
Licenciado como Doutor em Quiropraxia (DC) em Texas & Novo México*
Licença DC do Texas # TX5807, Novo México DC Licença # NM-DC2182
Licenciada como enfermeira registrada (RN*) in Florida
Licença da Flórida Licença RN # RN9617241 (Controle nº 3558029)
Status compacto: Licença Multiestadual: Autorizado para exercer em Estados 40*
Alex Jimenez DC, MSACP, RN* CIFM*, IFMCP*, ATN*, CCST
Meu cartão de visita digital





